

Alpha1-antitrypsin deficiency (AATD) is a well known genetic risk factor for pulmonary disease and is the most frequent hereditary disease diagnosed in adults. Despite being one of the most common hereditary diseases, AATD remains under-diagnosed because of its variable clinical presentation and the poor knowledge of this disease by physicians. With the aim of identifying clinical differences that could influence early diagnosis, we compared two groups of six AATD Pi*ZZ patients with different lung function severity and clinical expression at diagnosis. On comparing the two groups, we observed a younger mean age at diagnosis and more exacerbations in the severe group, but the percentage of smokers did not statistically differ between the two groups. Our results suggest that AATD continues being a disease suspected on younger patients with a worse lung function. In addition these findings confirm the clinical variability of the disease and that there are still unknown factors that contribute to its development. Therefore, early diagnosis may modify the prognosis of this disease.
Alpha1-antitrypsin deficiency (AATD) is a well known genetic risk factor for pulmonary disease and is the most frequent hereditary disease diagnosed in adults. AATD has a prevalence of less than 5 cases per 10,000 habitants, accounting for 1–2% of patients with emphysema.1,2 Despite being one of the most common hereditary diseases, AATD remains under-diagnosed because of its variable clinical presentation and the poor knowledge that physicians have of this disease. Tobacco exposure is the most important risk factor for the development of emphysema in AATD.3,4
Although forced expiratory volume in the first second (FEV1) and diffusing lung capacity for carbon monoxide (DLCO) are the usual parameters used in clinical practice to follow disease progression,5 their great variability makes them less suitable to be used as outcomes in clinical trials.6 Loss of lung density measured by quantitative computed tomography has demonstrated to be a more accurate outcome measure7–9 and has a good long-term correlation with FEV1, DLCO, and survival.10,11
Despite the data reported from patient registries, the evolution of lung disease associated with AATD is not fully understood.12,13
Since this is a progressive disease, early diagnosis and treatment may change the natural course of the disease.
With the aim of identifying clinical differences that could influence early diagnosis, we consecutively selected patients from our database of AATD patients, and we compared two groups according to the severity of the disease. Severe disease defined as FEV1<45% at diagnosis and then we found patients with FEV1>65% paired by age and gender.
Clinical observationsModerate groupSix AATD Pi*ZZ patients with relatively preserved lung function at diagnosis (FEV1≥65%) were included. The mean age at diagnosis was 51 years (SD=4) and four of patients were men. Two patients were never smokers and 4 were former smokers. The mean FEV1 at diagnosis was 93% (SD=29). Regarding comorbidities, one patient had bronchiectasis, another cardiovascular disease and one had severe liver disease requiring liver transplantation. None of the six patients had had more than one exacerbation during the previous year. Two patients had radiologically confirmed emphysema with a FEV1 of 65 and 67% respectively and intravenous augmentation treatment was initiated after diagnosis (Table 1).
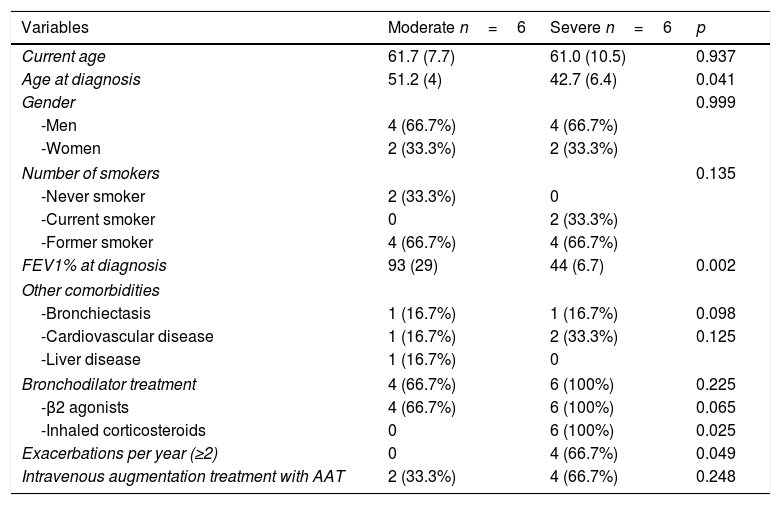
Demographic and clinical characteristics.
| Variables | Moderate n=6 | Severe n=6 | p |
|---|---|---|---|
| Current age | 61.7 (7.7) | 61.0 (10.5) | 0.937 |
| Age at diagnosis | 51.2 (4) | 42.7 (6.4) | 0.041 |
| Gender | 0.999 | ||
| -Men | 4 (66.7%) | 4 (66.7%) | |
| -Women | 2 (33.3%) | 2 (33.3%) | |
| Number of smokers | 0.135 | ||
| -Never smoker | 2 (33.3%) | 0 | |
| -Current smoker | 0 | 2 (33.3%) | |
| -Former smoker | 4 (66.7%) | 4 (66.7%) | |
| FEV1% at diagnosis | 93 (29) | 44 (6.7) | 0.002 |
| Other comorbidities | |||
| -Bronchiectasis | 1 (16.7%) | 1 (16.7%) | 0.098 |
| -Cardiovascular disease | 1 (16.7%) | 2 (33.3%) | 0.125 |
| -Liver disease | 1 (16.7%) | 0 | |
| Bronchodilator treatment | 4 (66.7%) | 6 (100%) | 0.225 |
| -β2 agonists | 4 (66.7%) | 6 (100%) | 0.065 |
| -Inhaled corticosteroids | 0 | 6 (100%) | 0.025 |
| Exacerbations per year (≥2) | 0 | 4 (66.7%) | 0.049 |
| Intravenous augmentation treatment with AAT | 2 (33.3%) | 4 (66.7%) | 0.248 |
Data are expressed as mean and percentages.
Six AATD Pi*ZZ patients with impaired lung function at diagnosis (FEV1≤45%) were analyzed. The mean age was 43 years (SD=6.4) and four were men. Two patients were current smokers and 4 were former smokers. The mean FEV1 at diagnosis was 44% (SD=6.7). We observed bronchiectasis in one patient and cardiovascular disease in two patients and none presented significant liver disease. All had had two or more exacerbations during the last year. Four patients initiated intravenous augmentation treatment after diagnosis, the two remaining patients were active smokers and therefore augmentation treatment was not indicated.
DiscussionDiagnosis of lung disease in AATD is usually delayed for some years after the first clinical manifestations.14 We compared two groups of patients diagnosed with lung disease of different severity to try to understand factors associated with early diagnosis.
On comparing the two groups, we observed a younger mean age at diagnosis and more exacerbations in the severe group, but the percentage of smokers did not statistically differ between the two groups.
Our results suggest that AATD continues to be a disease which is suspected in younger patients with a worse lung function. In addition these findings confirm the clinical variability of the disease and that there are still unknown factors that contribute to its development. Therefore, early diagnosis may modify the prognosis of this disease. It is estimated that a total of 253,404 ZZ individuals are distributed mainly in Europe and North America.2 Nevertheless AATD is under-diagnosed because of the poor knowledge of this disease by physicians. Indeed, in the study by Greulich et al. only 18 and 25% of physicians in Italy and Germany, respectively, stated that they tested all their patients with chronic obstructive pulmonary disease (COPD) and asthma.15 Likewise, in another survey conducted by Esquinas et al. in Spain and Portugal only 15.8% of physicians tested all of their COPD patients for AATD, and only 45.2% knew the AAT serum concentrations needed to consider severe deficiency.16
These data are in contrast with the worldwide recommendation of testing all COPD patients at least once in their lives.1,17,18 A population-based study performed in primary care in our region demonstrated that requests for AAT determinations were low compared to the number of COPD patients diagnosed per year.19 In addition, an important number of determinations were not prescribed because of pulmonary disease.20
ConclusionIn conclusion, lung disease associated with AATD may evolve in different ways and predisposing factors are not completely understood, which makes its early diagnosis even more important. Early diagnosis would help to improve strategies such as tobacco cessation, vaccination, initiation of optimum COPD treatment and augmentation therapy with AAT when indicated, to prevent disease progression. In addition, physicians should have a broader knowledge of the disease which would facilitate early diagnosis and thereby implement more efficient health care interventions.
Conflicts of interestMarc Miravitlles has received speaker or consulting fees from (in alphabetical order) Bial, Boehringer Ingelheim, Chiesi, Cipla, CSL Behring, Laboratorios Esteve, Gebro Pharma, GlaxoSmithKline, Grifols, Menarini, Mereo Biopharma, Novartis, pH Pharma, Rovi, TEVA, Verona Pharma and Zambon, and research grants from GlaxoSmithKline and Grifols.


