


Idiopathic pleuroparenchymal fibroelastosis (IPPFE) was recognized as a rare new entity. We report the case of a 63 years old female suffering from progressive dyspnea and dry cough for three years. Two years before admission to our hospital, idiopathic pulmonary fibrosis (IPF) was diagnosed in another hospital and treatment with prednisolone and N-acetylcysteine (NAC) was commenced. At admission HRCT showed upper lobe dominant fibrosis and associated pleural thickening. Surgical biopsies were re-evaluated and revealed fibroelastosis with pleural thickening and a probable UIP pattern, consistent with idiopathic PPFE. Treatment with pirfenidone was initiated due to progression under prednisolone and NAC. Upper lobe predominant pleural thickening with associated subpleural fibrotic changes should raise suspicion of PPFE.
Pleuroparenchymal fibroelastosis (PPFE) is a rare entity, characterized by upper lobe dominant subpleural fibroelastosis and dense fibrous thickening of the visceral pleura. Most cases are idiopathic. Evidence based treatment options do not exist.1 In the update of the international consensus classification of idiopathic interstitial pneumonia (IIP), idiopathic PPFE has been included within the group of rare IIP.2,3 We here report a case of PPFE, which was initially misdiagnosed as IPF.
Case presentationA 63 year old female patient presented with progressive dyspnea and dry cough for 3 years. There was no significant occupational exposure, allergy or smoking history. There was no family history of pulmonary fibrosis.
Two years before the admission to our hospital high-resolution computed tomography (HRCT) in a regional hospital had shown fibrotic changes of the lung parenchyma. Subsequently the patient underwent a video assisted thoracoscopic surgery (VATS) for histological assessment, fibrotic changes were interpreted as normal interstitial pneumonia (UIP) pattern. A diagnosis of IPF was made and a treatment with prednisolone and NAC started.
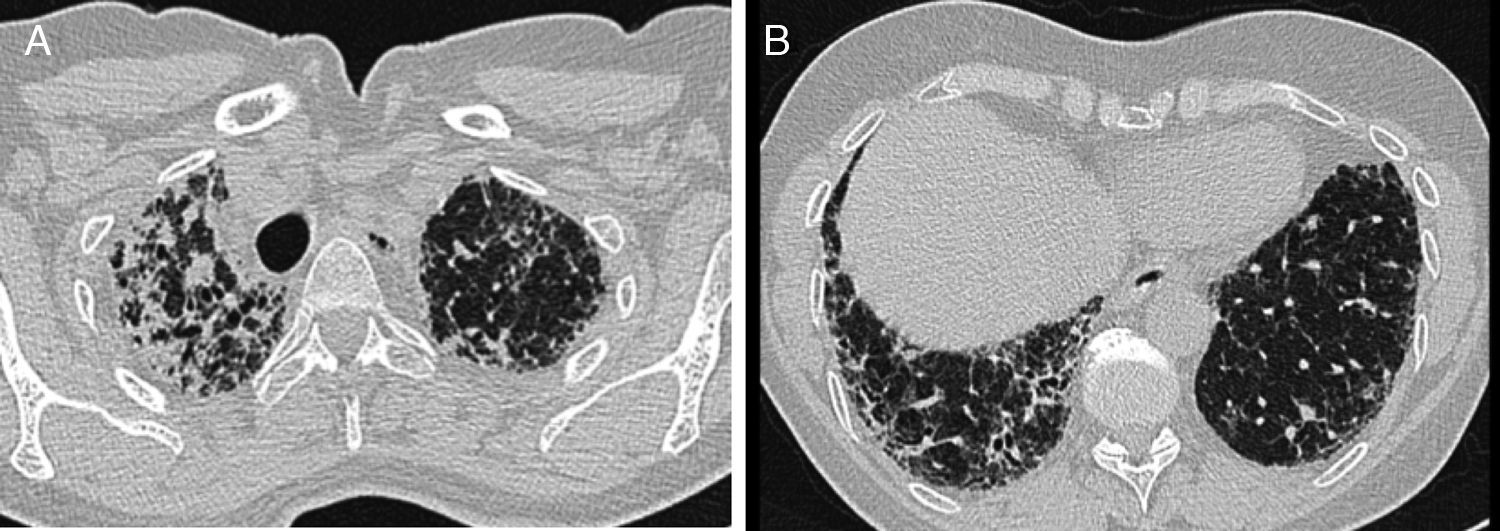
HRCT performed in our hospital demonstrated upper lobe predominant pleural thickening and associated reticular opacities, traction bronchiectasis, subpleural and peribronchial consolidation. There was upper lobe shrinkage and architectural distortion (Fig. 1A). The lower lobes showed minor honeycombing (Fig. 1B). Bronchoalveolar lavage cell differential was normal. There was no clinical or serological evidence of a connective tissue disease, vasculitis or extrinsic allergic alveolitis. Lung function showed a significant decrease in forced vital capacity (FVC) (55% of predicted) in comparison to two years earlier (72% of predicted) (Table 1). Six-minute walking test demonstrated a walking distance of 475m with significant desaturations (76%). Transthoracic echocardiography showed mild pulmonary hypertension with an estimated systolic pulmonary artery pressure of 35mmHg.
 Pleural thickening, dense fibrosis and consolidation in the upper lobes. (B) Traction bronchiectasis, reticular opacities and minor honeycombing in the lower lobes.")
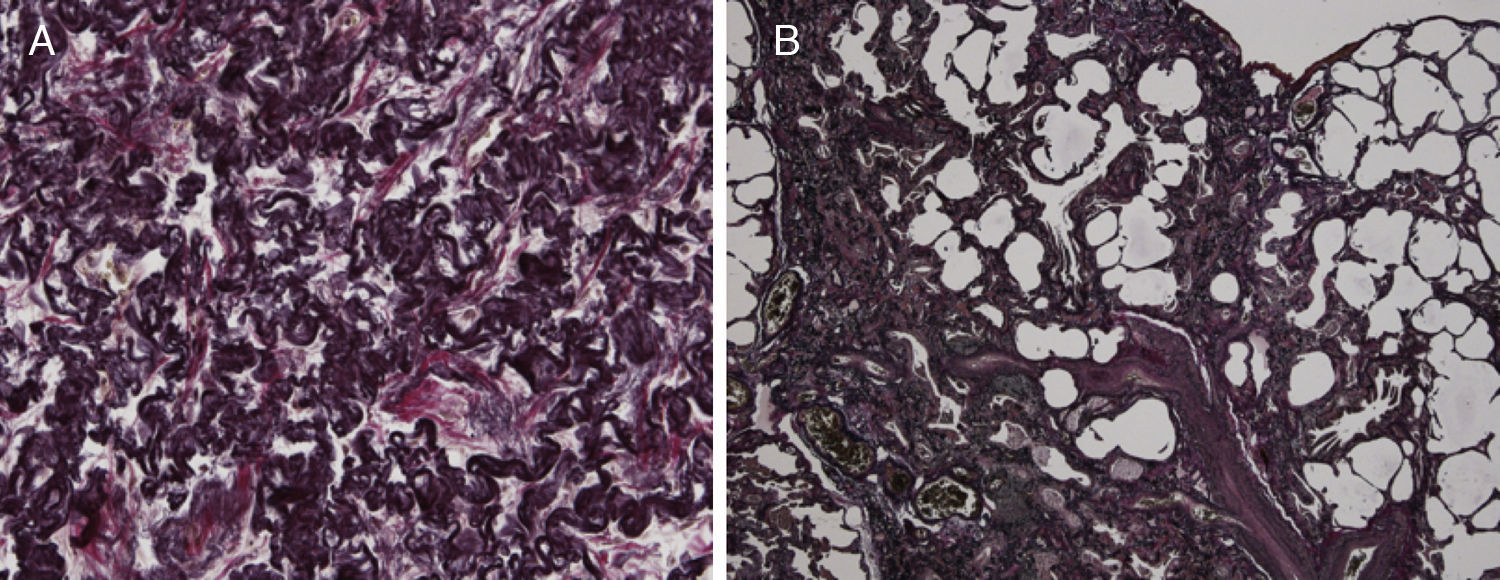
Because of the atypical location of the fibrotic changes predominantly in the upper lobes and the marked pleural thickening, histological specimens were reassessed in our pathology department. Histologic re-examination revealed the characteristic findings of PPFE with significant subpleural elastosis and alveolar-septal fibrosis in the left upper lobe (Fig. 2A) and a probable UIP pattern without fibroblastic foci in the lingula (Fig. 2B).
 The biopsy from the upper lobe showed severe fibrosis in the subpleural space with a prominent elastosis in the EvG stain; the adjacent pulmonary parenchyma revealed non-characteristic alveolar-septal fibrosis without honeycombing. (B) The biopsy from the lingular lobe revealed a patchy fibrosis with paraseptal and subpleural predominance. Honeycombing and mucus plugging were seen focally. Fibroblastic foci were not detected. Inflammatory infiltrates were scarcely developed. In some parts proliferation of smooth muscle cells was found. A mesh-like fibrosis was seen focally.")
(A) The biopsy from the upper lobe showed severe fibrosis in the subpleural space with a prominent elastosis in the EvG stain; the adjacent pulmonary parenchyma revealed non-characteristic alveolar-septal fibrosis without honeycombing. (B) The biopsy from the lingular lobe revealed a patchy fibrosis with paraseptal and subpleural predominance. Honeycombing and mucus plugging were seen focally. Fibroblastic foci were not detected. Inflammatory infiltrates were scarcely developed. In some parts proliferation of smooth muscle cells was found. A mesh-like fibrosis was seen focally.
Due to the marked lung function decline and the progression of dyspnea over 3 years, we started treatment with pirfenidone. Long term oxygen treatment was also started because of significant desaturation on exertion. After 6 and 12 months of treatment with pirfenidone FVC and diffusing capacity of the lung for carbon monoxide (DLCO) further declined and the treatment was stopped. The patient was referred for evaluation for lung transplantation, however due to the comorbidity of severe osteoporosis and advanced age, lung transplantation was denied. The patient died 9 months later from progressive respiratory insufficiency.
DiscussionIdiopathic PPFE was first described by Amitani et al. as idiopathic pulmonary upper lobe fibrosis.4 Frankel et al. coined the term idiopathic PPFE for this new clinico-pathological entity.5 In the updated IIP classification of 2013, PPFE was recognized as a distinct disease and was included in the rare IIP subgroup.2
The entity was initially described by Japanese groups from 19926 onwards, later in other countries in small case series.5,7–9 Reddy et al. presented data of 12 patients as well as a meta-analysis of 39 patients with PPFE from the literature.1 Regarding etiology autoimmunity, familial and genetic factors have been described. Camus et al. reviewed secondary PPFE induced by chemotherapy.10 Further cases have been reported secondary to bone marrow transplantation as well as lung transplantation.11,12 Interestingly, the complication rate of pneumothorax and pneumo-mediastinum is higher in PPFE than in IPF.13 The differential diagnosis is fibrotic sarcoidosis, tuberculosis, asbestosis, connective tissue diseases, and pleural fibrosis after radiation therapy or after coronary bypass operation.7,10
In our case the histopathological changes in the lingular lobe could be classified as probable UIP pattern, whereas the upper lobe changes were consistent with PPFE. Reddy et al. described concurrent patterns of PPFE with UIP in 25% of their patients. The relevance of coexisting different patterns in terms of prognosis is not clear at present. Survival of PPFE patients tends to be shorter than in IPF.13
In our case of PPFE with coexisting UIP pattern we started treatment with pirfenidone because of progression under combined prednisone and NAC therapy after a discussion in a multidisciplinary meeting as an off-label individual treatment. Pirfenidone is currently only approved for the treatment of IPF.14 There are limited data about its efficacy in other fibrotic lung diseases. Recently 5 patients with connective tissue disease associated interstitial lung disease (CTD-ILD) were treated with pirfenidone and all showed functional improvement.15 A randomized controlled trial is currently ongoing in Germany to assess the efficacy of pirfenidone in non-IPF fibrosis (EudraCT 2014-000861-32 3).
In our patient, the treatment with pirfenidone with standard daily dosage (2403mg/day) was initially well tolerated; however there was no improvement in dry cough and dyspnea. The treatment was stopped after one year due to significant decline in lung function (more than 10% of FVC) and 8kg of weight loss. There are only sporadic reports on PPFE treatment, including antifibrotic drugs.9,12 The collection of further evidence for diagnostic criteria and treatment of such a rare disease is challenging but necessary.
In conclusion, our case of idiopathic PPFE with coexisting UIP pattern was first misdiagnosed as IPF. It is important to consider PPFE as a differential diagnosis in every patient with ILD and upper lobe dominant pleural thickening with adjacent subpleural fibrosis. This case confirms the value of the multi-disciplinary discussion as a reliable diagnostic tool for differential diagnosis of ILD subtypes.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.


