

Dear editor
Alpha-1-antitrypsin (AAT) is the major protease inhibitor in serum whose function is the control of neutrophil elastase.1-3
AAT deficiency (AATD) is an autosomal codominant disorder caused by SERPINA1 mutations4 increasing the risk of emphysema and chronic obstructive pulmonary disease (COPD). Despite most cases being connected with low AAT concentrations and PI*ZZ and PI* SZ genotypes, in rare instances, those can be correlated with rare deficiency or null variants.4 Until now, more than 120 SERPINA1 mutations were reported and in Portugal 9.5% of patients were linked to null alleles.2,5
Here, we present a 46-year-old woman, former smoker (37 pack-per-year), with no family history of lung disease or occupational exposure. She was referred to our center, in 2016, with Asthma-COPD diagnosis and a mild form of AATD as indicated by a PI*MZ genotype obtained in 2012. The patient described a 4-year history of exertional dyspnea (mMRC3), wheezing and cough with frequent minimal sputum. Physical examination showed decreased breath sounds in both lungs. Patient was medicated with formoterol 24 µg, ipratropium bromide 80 µg and, aminophylline 200 mg twice a day and salbutamol 100 µg as needed with poor symptomatic control and at least one moderate to severe exacerbation per year. Chest computed (CT) tomography confirmed pulmonary hyperinflation with centrilobular and panlobular emphysema. A forced expiratory volume (FEV1)/forced vital capacity (FVC) ratio of 53% and a FEV1 of 60% of the predicted value (1.52 L) were estimated (GOLD 2, Group B). Body plethysmography showed elevation in residual volume (RV: 3.13 L; 217% of predicted) and diffusion capacity of carbon monoxide (DLCO) was moderately decreased (45% of predicted). Arterial blood gas showed hypoxemia (pO2 66 mmHg). AAT concentration was 5 mg/dl as measured by nephelometry. Given that patient´s symptoms and AAT levels were inconsistent with a PI*MZ result, the genetic study was repeated in an independent laboratory.3
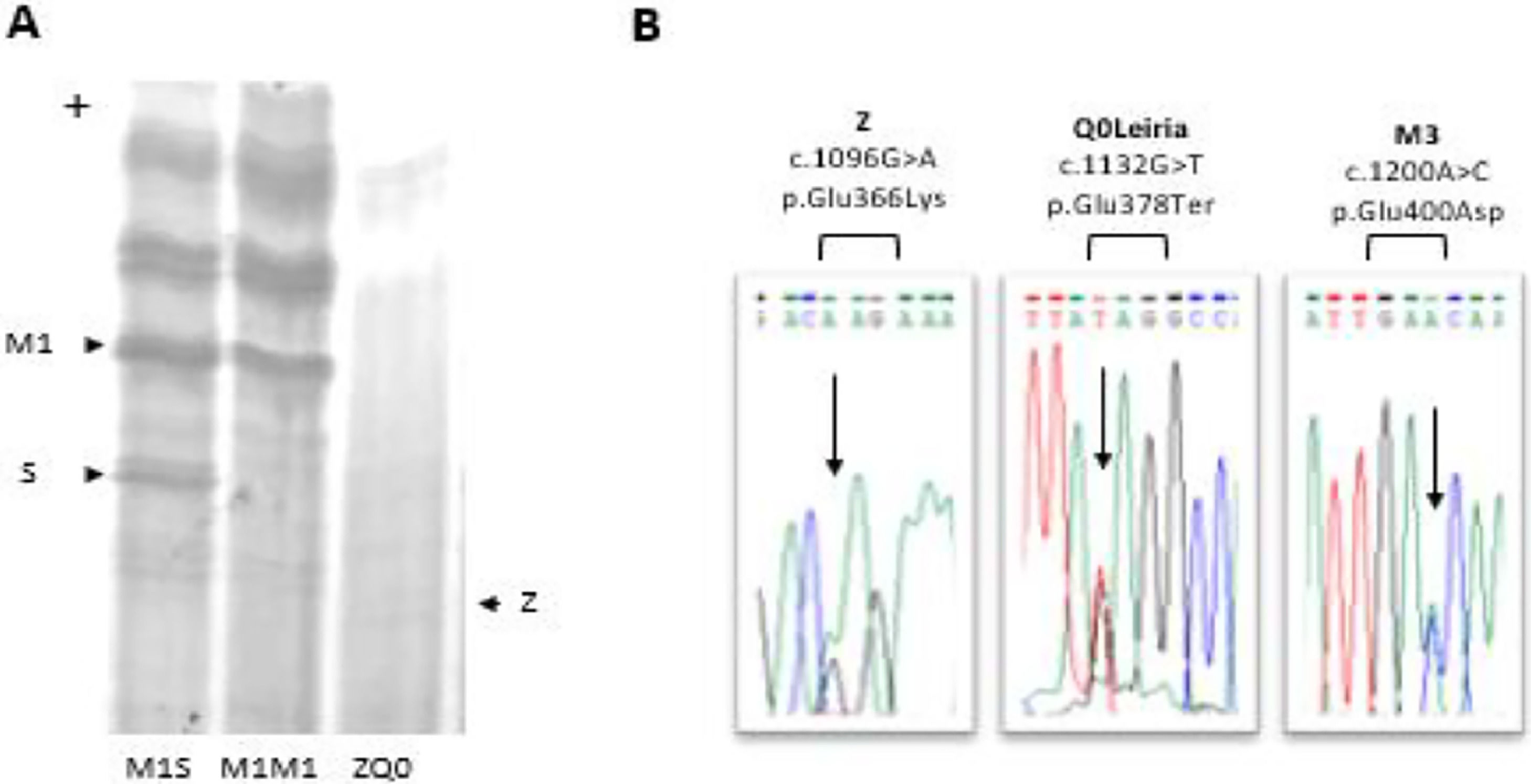
A PI*ZQ0 diagnosis was achieved, where the null allele was caused by novel mutation leading to the premature termination of protein sequence (NM_000295.5: c.1132G>T; p.Glu378Ter). This mutation found to be associated with a PI*M3 (p.Glu400Asp) background and was labeled as Q0Leiria according to AATD nomenclature (Fig. 1).
 Protein gel electrophoresis. Index case ZQ0 displays only a band corresponding to PI*Z allele. (B) Electropherogram of the index case for SERPINA1 (NM_000295.5) exon 5, covering the mutations that define Z, Q0Leiria and M3 alleles. The arrows show the position of the corresponding nucleotide substitutions.")
Characterization of ZQ0Leiria index case. (A) Protein gel electrophoresis. Index case ZQ0 displays only a band corresponding to PI*Z allele. (B) Electropherogram of the index case for SERPINA1 (NM_000295.5) exon 5, covering the mutations that define Z, Q0Leiria and M3 alleles. The arrows show the position of the corresponding nucleotide substitutions.
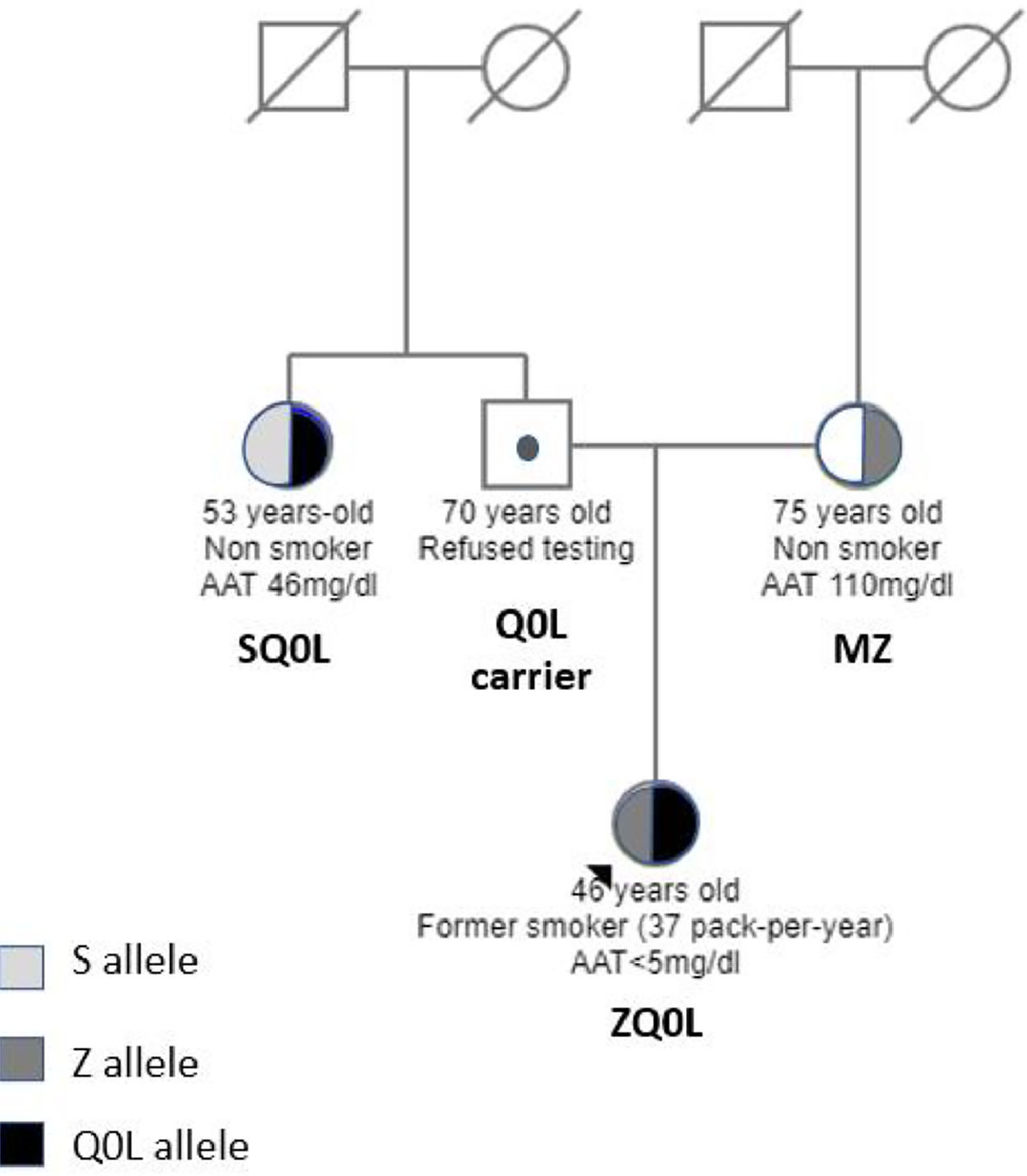
The family screening uncovered that her mother (75-year-old, nonsmoker) had a PI*MZ genotype with 110 mg/dl of AAT. No evidence of lung disease was detected (normal CT and lung function tests). Her 53-year-old aunt (non-smoker) had a PI*SQ0Leiria genotype with 46 mg/dl of AAT without respiratory symptoms or evidence of disease (normal CT and lung function tests) (Fig. 2).

Unfortunately, we lost the follow-up of this patient in 2017 and no AAT therapy could be implemented in our pulmonology department.
The Q0Leiria allele resulting from a premature termination of protein 41 codons upstream of the wildtype impairs AAT secretion as indicated by carriers low AAT concentrations and absence of a corresponding band in gels. To our knowledge this mutation is not described in the literature, nor is it present in large genomic databases.
Null mutations as Q0Leiria are not associated with AAT polymerization in the liver nor in the lung interstitium and thus, no exacerbation of pulmonary disease is expected once their clinical manifestations result exclusively from AAT loss of function. The rarity of these alleles impairs a proper evaluation of their disease risk when combined with known deficiency alleles.6
Although both cases analyzed in this report show AAT values bellow the protective threshold of 57 mg/dl only the smoker PI*ZQ0Leiria carrier presents her lung function seriously compromised. This confirms the importance of tobacco smoking on AATD clinical presentations. Indeed, smoke exposure is a well-known risk factor for the onset and progression of Emphysema and COPD without which severe AATD cases such the one of PI*SQ0Leira may not show symptoms of pulmonary disease.6 Similar findings were reported for never-smokers with PI*SQ0Ourém genotypes.7
This report also highlights the importance of a prompt diagnosis and the need to access AAT concentration together with a detailed clinical evaluation. Without the possibility of performing the genetic test in a reference laboratory offering an algorithm of diagnosis (AAT phenotyping, genotyping and SERPINA1 sequencing) these subjects would persist misdiagnosed. The detection of Null alleles should lead to genetic counseling and a possible use of replacement therapy.


