


Cystic fibrosis (CF OMIM: #219700) is an autosomal recessive disorder caused by pathogenic variants in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator).1 Among the 2,106 variants described in CFTR, large deletions or insertions are considered rare [59 (2.80%)].2 The identification of large alterations in the CFTR is challenging and might result in wrong diagnosis, indicating false-negative in carriers of rare variants that are potentially severe.3,4 Thus, the implementation of additional techniques in the CF diagnosis workflow becomes necessary, which includes the use of Multiplex Ligation Probe Amplification (MLPA) to identify chromosome rearrangements, deletions, and insertions.3 So we aimed to describe the genetic profile of large deletions or insertions in CFTR identified using MLPA and to describe its influence on CF patients’ phenotype in a referral center.
Five CF patients (chloride over 60 mEq/L in two sweat tests) presenting at least one pathogenic variant in the CFTR characterized as a large deletion or insertion were included after the study approval by the Ethics Committee (#78192216.2.0000.5404). The caregivers of the CF patients who participated in our study signed the consent to publish patients’ data. The screening of the pathogenic variants in the CFTR was carried out as previously desbribed.5 The following markers were described: patients’ age at diagnosis; ethnic group; spirometry, classified according to the forced expiratory volume (FEV1) at different levels of obstruction: mild (≥70%), moderate (60-69%), moderately severe (50-59%), severe (35-49%), and very severe (<35%); Shwachman-Kulczycki score graded as excellent (86-100), good (71-85), mild (56-70), moderate (41-55), and severe (≤40)6; immunoreactive trypsinogen; and sweat test results. The microbiological evaluation was carried out for the colonization by 11 microorganisms. In addition, the comorbidities and medication used by the patients were described.
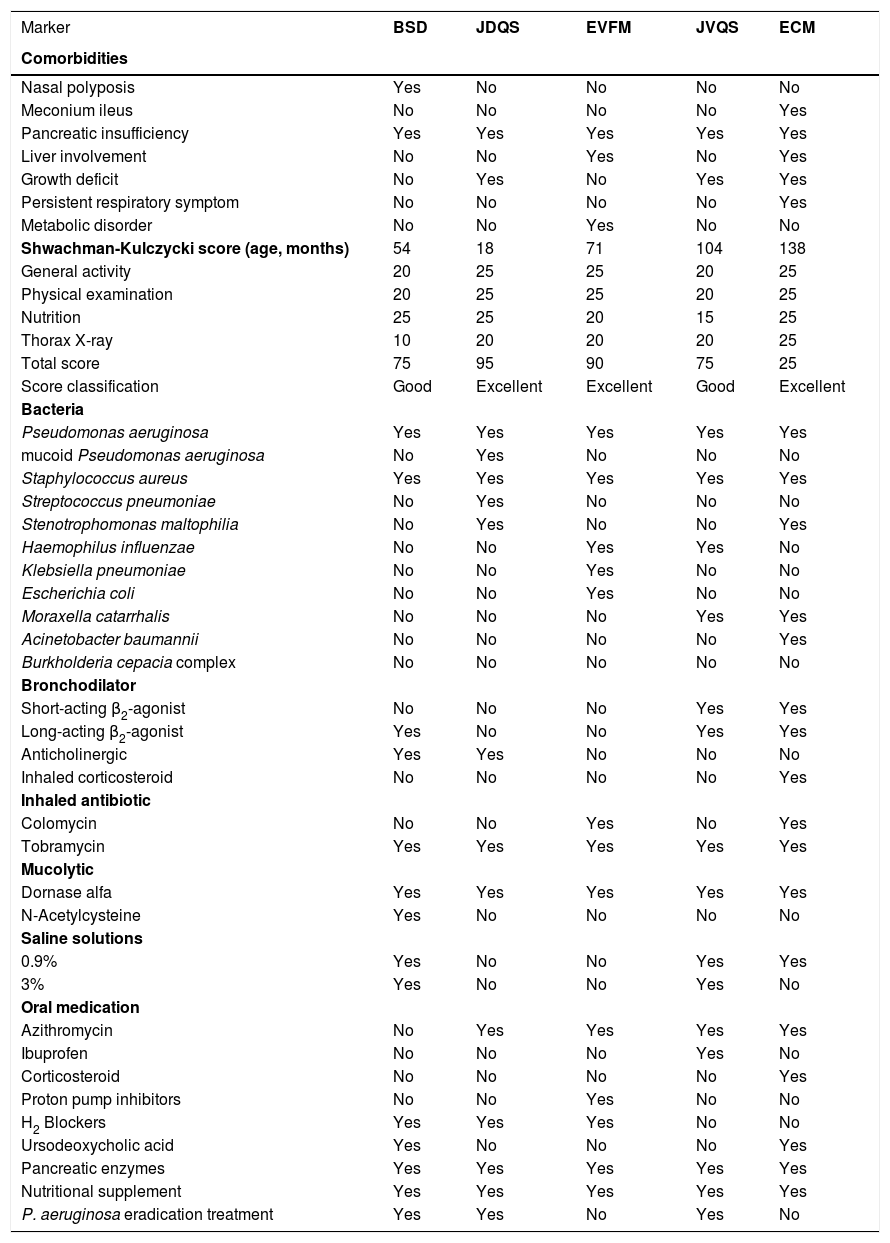
All the patients had one identified variant, c.1521_1523delCTT (F508del; p.Phe508del). The MLPA technique also identified four variants considered large deletions or insertions, namely, CFTRdele7-18, CFTRdup6b-16, CFTRdele14b+CFTRdup9, and CFTRdele16-20. The variant CFTRdele16-20 was identified in two patients. The CFTR genotype, race, and diagnostic tests were described (Table 1), as well as the comorbidities, Shwachman-Kulczycki score, microorganism profile, and medication used by the patients (Table 2).
Description of genotype, race, and diagnostic tests results in cystic fibrosis patients in the presence of CFTR large deletions or insertions.
| Marker | BSD | JDQS | EVFM | JVQS | ECM | |
|---|---|---|---|---|---|---|
| CFTR Genotype | Allele 1 | F508del | F508del | F508del | F508del | F508del |
| Allele 2 | CFTRdele7-18 | CFTRdele16-20 | CFTRdup6b-16 | CFTRdele16-20 | CFTRdele14b and CFTRdup9 | |
| Race | Caucasian | Caucasian | Caucasian | Mixed race | Caucasian | |
| IRT (ng/mL)* | 204/131 | 86/138 | 262/354 | 159/147 | 260/410 | |
| Sweat chloride ion (mEq/L)* | 89/92 | 95/92 | 95/90 | 110/106 | 116/128 |
F508del; p.Phe508del = c.1521_1523delCTT; IRT: immunoreactive trypsinogen; CFTR: Cystic Fibrosis Transmembrane Regulator.
Comorbidities, Shwachman-Kulczycki score and medications in cystic fibrosis patients in the presence of CFTR large deletions or insertions.
In our cohort, four patients were self-declared Caucasians, and one was of mixed race; four of them were female. Two patients were diagnosed when they were five months old; two were two months old; and one was one month old. The Shwachman-Kulczycki score varied distinctly for each participant. All participants were colonized by Pseudomonas aeruginosa and Staphylococcus aureus, while unequal colonization by other microorganisms was observed in the patients. All participants used inhaled antibiotics, mucolytic agents, nutritional supplements, and pancreatic enzymes; four patients used bronchodilator and one used inhaled corticosteroid. In addition, all patients in our study cohort had pancreatic insufficiency (Table 2).
Since the CFTR pathogenic variants present different effects on the phenotype, it seems relevant to optimize the detection method to avoid inaccurate and/or delayed diagnosis.7,8 In such contexts, the MLPA technique implementation in the CF diagnosis should be optimized.7 For instance, Atag et al. (2019) evaluated 250 CF patients that presented 80 genetic distinct variants in the CFTR and, out of those, five (CFTRdele2, CFTRdele4-11, CFTRdele5-10, CFTRdele12, and CFTRdele19-21) were characterized as large deletions and occurred in 16 CF patients. Large deletions were associated to the worst pulmonary phenotype, pancreatic insufficiency and liver involvement.8 The same findings were reported by Martins et al. (2019) who reported the presence of a severe phenotype with pancreatic insufficiency and infection by P. aeruginosa9 in five patients with large deletions or insertions in the CFTR.
The identification of all types of CFTR variants, including large deletions and insertions, should be one of the main points to be considered in the patients’ differential diagnosis.4 For example, in a study carried out in Serbia, twenty-two different CFTR variants were identified in the population studied, evidence of high heterogeneity. Most of these variants had not been reported in neighboring countries, possibly due to the use of commercial tests for CF diagnosis in those places, which did not include the MLPA technique. Due to the use of different molecular analysis techniques, an increase from 54.45% to 72.8% was observed in the effectiveness rate to identify the CFTR genotype.10
The description of clinical manifestations along with the identification of large deletions or insertions in the CFTR pointed out a more severe phenotype of these patients in our serial case report. And, although younger patients do not present some symptoms, there is still great potential for developing them in the future. Currently, there is no corrective therapy for CFTR large deletions or insertions, due to the difficulties of modulating the impact of these large deletions and insertions in the gene expression mechanisms.11
In conclusion, our study identified four genetic variants of the type CFTR large deletions and insertions, which were characterized by their low genotypic and diagnostic frequency. Two participants presented the same variant, while the variants identified in the other three participants were unique. The identification of large deletions and insertions through a broader genetic analysis is very important for CF diagnosis, since those variants, despite being rare, might be associated with the disease higher severity phenotypes.
Author contributionAll authors approved the manuscript and agreed with its submission to the journal. Also, all authors wrote and revised the manuscript.
The pediatrics outpatient service of the University Hospital for collaborating with the data collection.


