









In recent decades, the classification of idiopathic interstitial pneumonias (IIPs) has been based on clinical, radiological, and histopathologic features; this approach aims to identify entities with more or less specific and predictable clinical behaviour.1 However, this process of phenotype-based identification cannot properly distinguish idiopathic entities from similar conditions that have already identified causes or triggers, and does not provide clues to identify efficacious treatment approaches.1
The introduction of molecular morphology analyses, single-cell RNA sequencing, and advances in the knowledge of genetic and molecular backgrounds have greatly contributed to the identification of the pathobiological pathways underlying these disorders and potential endotypes. A mechanistic-based approach that employs the precision medicine concepts similar to oncology has progressively enabled the development of new and more efficacious therapeutic strategies and yielded changes in the prognoses.2 Since the current ATS/ERS classification system1 represents a milestone of the clinical work-up of interstitial lung diseases (ILDs), we aimed to focus on the main and, somehow common, pathogenetic backgrounds of IIPs and similar entities with known causes or triggers to identify the main ‘pathogenetic categories’. By analysis of the pathogenetic processes of ILDs, we carried out the suggestions for four pathogenetic proposed categories; the first one includes entities showing senescence as a pivotal step and a clinical trajectory similar to that observed in idiopathic pulmonary fibrosis (IPF, which is mainly a subset of fibrosing hypersensitivity pneumonitis [HP] and a subset of collagen vascular diseases-ILDs); the second category encompasses all disorders with a potential fibrotic evolution but with a clonal background; the third category has inflammation as a pivotal factor with a clinical behaviour potentially modifiable by anti-inflammatory-immunomodulating drugs; the last one coveres all the monogenic entities in which senescence/alveolar stem cell exhaustion is not the main pathogenetic trait. Table 1 presents the above-mentioned categorisation scheme. The current article is the first of four narrative reviews. For this specific narrative review, three of the authors searched in PubMed with the following final search string, reached by consensus: (‘idiopathic pulmonary fibrosis/classification’ [MeSH Terms] OR ‘idiopathic pulmonary fibrosis/diagnosis’ [MeSH Terms] OR ‘idiopathic pulmonary fibrosis/aetiology’ [MeSH Terms] OR ‘idiopathic pulmonary fibrosis/pathology’ [MeSH Terms] OR ‘idiopathic pulmonary fibrosis/physiopathology’ [MeSH Terms]). We also performed a search using the terms ‘pathogenesis’, ‘basal cells’, ‘honeycombing’, ‘immunohistochemistry’, and ‘RNA sequencing’.
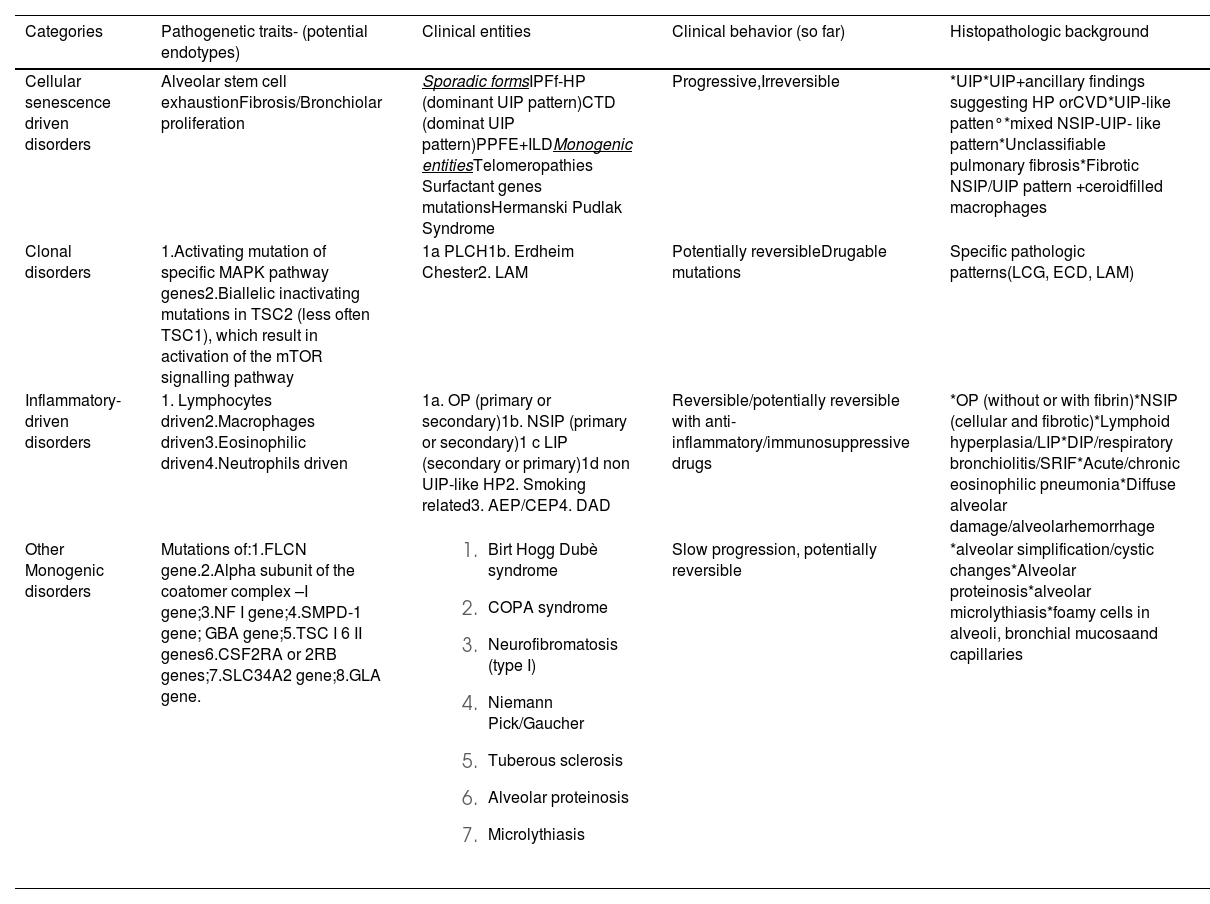
Pathogenetic categories of Idiopathic Interstitial Pneumonias and akin disorders.
1. IP secondary to cell senescence/alveolar stem cell exhaustion/fibrosis/bronchiolar proliferation
This category includes chronic/fibrotic ILDs independent of their putative etiological features. These diseases are usually radiologically and histologically characterised by the usual interstitial pneumonia (UIP) pattern.1 The rationale for this proposal is based on the emerging awareness that the prognosis of progressive ILDs (PILDs) and their response to available antifibrotic therapies mainly depends on the type and amount of activated or silenced structural and molecular pathways in the lung parenchyma more than their aetiology.3-10 These forms include IPF, familial pulmonary fibrosis (FPF), and a variety of fibrosing PILDs, as defined recently (mainly fibrosing and progressive HP and subsets of connective tissue disease [CTD]-ILDs).11-13 Although the UIP pattern is the most typical histological background in this group, it also includes cases or entities that may present without the typical UIP pattern.14 The pathogenetic processes occurring in this group can be summarised as follows: alveolar stem cell exhaustion, development of a micro-environmental alveolar fibrotic field, bronchiolisation, and honeycombing (Fig. 1).
 sparing the centrilobular region. Only one tongue of fibrotic tissue is “running” towards a bronchiole (triangle), The fibrotic process has an “arc-like” shape (line). (H&E,low power).")
Transbronchial cryobiopsy. The patchy fibrosis and scattered fibroblastic foci are evident along an interlobular septum (star) sparing the centrilobular region. Only one tongue of fibrotic tissue is “running” towards a bronchiole (triangle), The fibrotic process has an “arc-like” shape (line). (H&E,low power).
Defective/dysfunctional repair of alveoli is considered the primary pathogenic factor in IPF, and is centred on the loss of precursor cell function in type-II alveolar epithelial cells (AECII).15-17 This defect has also been documented in non-IPF fibrosing ILDs with a UIP pattern and in FPF, even without the typical UIP pattern (Table 1).18,19 AECII behave as an epithelial cell precursor within the alveolar niche, sharing complex interactions with mesenchymal cells. The latter are tightly regulated by the expression of signalling pathways such as the Wnt, transforming growth factor (TGF)-beta, Notch, and platelet-derived growth factor receptor pathways (Fig. 2).20-23 Chronic bidirectional perturbation of the crosstalk between epithelial and mesenchymal precursors is considered the initial pathogenic mechanism in IPF.24-28 This abnormal crosstalk, which represents the disease mechanicistic marker, was defined more precisely when genetic studies on FPF (and also sporadic cases) showed the specific gene mutations affecting the telomere length control or genes specifically expressed by AECII (surfactant proteins, ABCA).29-38 Thus, these observations provided an etiologic explanation of the early disease mechanisms centred on the progressive loss of stem/precursor reparative functions of AECII. The causes of this loss of function included senescence and unfolded protein response.38-46 Interestingly, senescence can be considered a stress response phenotype, thus reconciling the potential pathogenic contribution of different genetic abnormalities leading to stem cell exhaustion.47 Accordingly, IPF is characterised by the persistence of senescent transitional cells with a paucity of mature AECII, and the transitional state has a transcriptomic signature of cell cycle arrest.48,49
. AECII show nuclear beta-catenin (b) and the senescence-associated marker p16 (c). Honeycomb cyst: a continuous layer of early-fibrous tissue underlying the epithelial surface as demonstrated by tenascin (d) is covered by p16+ senescent epithelium (e). Basal cells in Honeycomb cyst show irregular distribution (f, DN-p63) and a senescent phenotype (g: p21). TBB3 clearly evidence both myofibroblasts and damaged epithelial cells (h). Basal express nuclear beta-catenin (i) and the WNT pathway target gene Cyclin-D1 (l).")
Histological and immunohistochemical analysis in UIP. Hyperplastic AECII cover densely the fibrotic interstititial tissue in the early alveolar lesions (a). AECII show nuclear beta-catenin (b) and the senescence-associated marker p16 (c). Honeycomb cyst: a continuous layer of early-fibrous tissue underlying the epithelial surface as demonstrated by tenascin (d) is covered by p16+ senescent epithelium (e). Basal cells in Honeycomb cyst show irregular distribution (f, DN-p63) and a senescent phenotype (g: p21). TBB3 clearly evidence both myofibroblasts and damaged epithelial cells (h). Basal express nuclear beta-catenin (i) and the WNT pathway target gene Cyclin-D1 (l).
The critical threshold of AECII senescence/stem cell exhaustion for triggering the pathologic process may be influenced by the concurrent action of different predisposing factors, including telomere attrition (occurring in ageing lungs), genetic predisposition (very robust in familial cases), toxic substances (e.g. cigarette smoke, asbestos, fine particles, drugs), and mechanical stress (explaining the peculiar anatomic location of early lesions in IPF).43,50-52 According to the available data, a variety of basic molecular mechanisms are also involved in a self-sustaining loop of aberrant signalling in IPF. These include epithelial-mesenchymal transition (EMT), aryl hydrocarbon receptor response, and autophagy.53-56 Further support for this pathogenic model is provided by clinical and experimental studies.57,58
Clinical/pathological message: The relevance of alveolar stem cell senescence highlights the potential role of these cells as the first and more relevant culprit in disorders showing the UIP pattern as the histopathologic background. Specific lung tissue or blood markers could be identified accordingly and used for diagnostic and prognostic purposes.
Abnormal cell signallingCell senescence is the permanent state of G1 arrest that limits the proliferation of damaged cells. Senescent AECII cannot sustain the proliferation and differentiation in the stem niche, resulting in the impairment of physiological homeostasis seen in the IPF alveoli.59 Senescent cells can eventually trigger the production of a secretome (the senescence-associated secretory phenotype [SASP]) that is enriched in proinflammatory cytokines, chemokines, reactive oxygen species, and growth factors and can alter the tissue micro-environment at involved sites.60-62 SASP signalling likely represents the main driver of the abnormal tissue remodelling in IPF and in fibrosing ILDs with a UIP-like background. It recruits bone marrow-derived mesenchymal stem cells and perturbs their differentiation. As a result, mesenchymal cell abnormalities are common in UIP and include the migration and activation of fibroblasts (myofibroblasts foci, scarring fibrosis), smooth muscle hyperplasia, deposition of elastin, ossification, fat metaplasia, and abnormal angiogenesis.63-66 In this complex scenario, the different molecular and cellular mechanisms involved include pathways regulating tissue development, morphogenesis and inflammation (Wnt, TGF-beta, Ras, signal transducer and activator of transcription proteins [STAT], etc.)28,67-72 (Fig. 3). In addition, the SASP effector pathway cGAS/STING may abnormally recruit macrophages and other immune cells,73,74-79 thus partly reconciling previous inflammation-centred models with more recent models of IPF pathogenesis.24,25,80-82
 provided by senescent AECII is able to trigger migration and deranged differentiation of mesenchymal stem cells causing myofibroblast accumulation and interstitial fibrosis. Due to senescence induced senescence (SIS) this process may trigger bronchiolar basal cell precursors affecting their differentiation. Abnormal basaloid cells may in turn provide pro-fibrotic signals to neighboring mesenchymal cells. This complex mechanism may severely affect the alveolar/bronchiolar junctions with eventual formation of honeycomb cysts, progressive remodeling and functional loss. DDR: DNA Damage Response; AECII: alveolar Epithelial Cells type-II; MSC: Mesenchymal Stem Cells; SASP: Senescence Associated Secretory Phenotype; SIS: Senescence Induced Senescence.")
Simplified scheme of IPF pathogenesis. The aberrant signaling (SASP) provided by senescent AECII is able to trigger migration and deranged differentiation of mesenchymal stem cells causing myofibroblast accumulation and interstitial fibrosis. Due to senescence induced senescence (SIS) this process may trigger bronchiolar basal cell precursors affecting their differentiation. Abnormal basaloid cells may in turn provide pro-fibrotic signals to neighboring mesenchymal cells. This complex mechanism may severely affect the alveolar/bronchiolar junctions with eventual formation of honeycomb cysts, progressive remodeling and functional loss. DDR: DNA Damage Response; AECII: alveolar Epithelial Cells type-II; MSC: Mesenchymal Stem Cells; SASP: Senescence Associated Secretory Phenotype; SIS: Senescence Induced Senescence.
Recent studies have demonstrated the potent fibrogenic contributions of monocytes and macrophages to IPF and experimental lung fibrosis, along with the mechanisms determining their recruitment.75,76,83-87 One of the main drivers of abnormal tissue remodelling is the propagation of senescence signalling in neighbouring bystander cells (senescence-induced senescence [SIS]).80-82 Cell senescence can be in fact transferred from the senescent to non-senescent neighbouring healthy cells by signals fuelled by senescent AECII (e.g. TGF-beta, Wnt) and by small extracellular vesicles derived from bronchiolar epithelial cells.83-87 Small extracellular vesicles can propagate senescence through miRNA cargo, a novel member of SASP, and represent new potential bio-markers and a therapeutic target in IPF.88-92 Furthermore, lung fibroblasts are senescent in IPF and are likely to contribute to further propagate senescence in their neighbourhood.93-96 A relevant effect of abnormal signalling is the survival of IPF fibroblasts through a mechanism related to mitochondrial dysfunction and autophagy impairment.97,98 The role of lymphatics and pleural/subpleural zones is not yet clear; however because this pro-fibrotic micro-environment is morphologically characterised by the presence of fibroblastic foci and areas of ‘patchy’ fibrosis having a periacinar distribution, these zones could be useful for inducing this process (Fig. 1).
Clinical/pathological message: Comprehension of the role of SASP acquisition and aberrant activation of an array of molecular pathways could lead to the development of new diagnostic and therapeutic options
HoneycombingHoneycombing (HC) is the most relevant diagnostic and prognostic marker in IPF and probably progressive pulmonary fibrotic ILDs.99-102 HC is characterised by modified distal bronchioles due to deranged epithelial/mesenchymal interactions that profoundly modify the differentiation of the alveolar-bronchiolar junction. This cascade involves EMT, mesenchymal stem cell recruitment, and waves of proliferation and post-mitotic senescence.59,103-111 The abnormal accumulation of Muc5b (a gel-forming mucin) related to promoter polymorphism within honeycomb cysts is likely implicated in mucociliary dysfunction.112-114 More recently, the role of fibrogenic ‘basal/basaloid’ cells has been recognised mainly in IPF.75,103,105,106,115-121,122 These cells, which are also referred to as basaloid cells, KR5-/KR17+ basal cells, abnormal airway basal cells, and senescent basaloid cells, have been detected and further characterised by single-cell RNA sequencing analyses, and have been shown to demonstrate a robust pro-fibrotic function that is partly mediated by the activation of macrophages110 (Fig. 4). Further studies are needed to clarify the origin of these aberrant basaloid cells (either modified airway basal cells or AECII-derived transitional basaloid cells).117,121,122 Gene expression profiles have been developed, suggesting that perturbation of micro-environmental niches in fibrotic lungs is mediated by discrete gene sets acting on fibroblasts and their precursors, including CTHRC1, a marker expressed by pathological myofibroblasts at fibroblast foci.123,124
![Micro-Honeycomb cysts: their aspect is characterized by “modified bronchioles” characterised by severely altered epithelial structure, fragmentation and disorganisation of the epithelial component as evidenced at CK5 immunostain for basal cells (a). The airway structure is severely compromised, with luminal epithelial cells fragmentation (b, H&E), loss of ciliated epithelium, and aphazard distribution of DN-p63+ basal cells (c). Nodular/elissoid alpha-SMA+ interstitial myofibroblast foci are common, occurring in segments of severe structural changes of the airway epithelium (d). These foci, previously described and named “Sandwich Foci” (SF) [Chilosi 2006;2013], are three-layered lesions formed by a CK5+ layer of basal/oid abnormal cells (e) interposed between superficial bronchiolar epithelial cells (often non-ciliated as evidence of bnormal differentiation) and the aSMA+ clusters of myofibroblasts (c). The basal/oid cells’ phenotype include the expression of molecules related to migratory activity as laminin-5-gamma-2 (f) and heath-shock protein-27 (g,h) as well as cell senescence (p16, i). These SF, that are fairly specific for the UIP pattern, may recapitulate major pathogenic mechanisms occurring in micro-Honeycomb cyst formation, and may also represent useful “biomarkers” in small cryobiopsy samples.](https://static.elsevier.es/multimedia/25310437/unassign/S2531043724000928/v1_202409160410/en/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w94UphxYc+GPca8Z7OggvdfJaI9Nqk/wRF1/+NWFHb4fNJcVZAnzxwqorc01lI58NvxkKFvxLpHwLhG5pQqIk4ROgQdH4os2VDyzUFWNfpZXf4F108xYWo+BiyIQ5MCVKWOZqOb++Gei5C0ynKULdQJGp+jiJhtw4b1WbZ2TGUfsUcCYo7d+05Y8HWkvSAzv5H+va4UfvZIebbixc7NiwFhIh6pjpXiS0Xv6cX1kpo8bshLl4sdLDkS/0wd1k+I5UZvUnsHEMrZXikTmnTjT/Wxc= "Micro-Honeycomb cysts: their aspect is characterized by “modified bronchioles” characterised by severely altered epithelial structure, fragmentation and disorganisation of the epithelial component as evidenced at CK5 immunostain for basal cells (a). The airway structure is severely compromised, with luminal epithelial cells fragmentation (b, H&E), loss of ciliated epithelium, and aphazard distribution of DN-p63+ basal cells (c). Nodular/elissoid alpha-SMA+ interstitial myofibroblast foci are common, occurring in segments of severe structural changes of the airway epithelium (d). These foci, previously described and named “Sandwich Foci” (SF) [Chilosi 2006;2013], are three-layered lesions formed by a CK5+ layer of basal/oid abnormal cells (e) interposed between superficial bronchiolar epithelial cells (often non-ciliated as evidence of bnormal differentiation) and the aSMA+ clusters of myofibroblasts (c). The basal/oid cells’ phenotype include the expression of molecules related to migratory activity as laminin-5-gamma-2 (f) and heath-shock protein-27 (g,h) as well as cell senescence (p16, i). These SF, that are fairly specific for the UIP pattern, may recapitulate major pathogenic mechanisms occurring in micro-Honeycomb cyst formation, and may also represent useful “biomarkers” in small cryobiopsy samples.")
Micro-Honeycomb cysts: their aspect is characterized by “modified bronchioles” characterised by severely altered epithelial structure, fragmentation and disorganisation of the epithelial component as evidenced at CK5 immunostain for basal cells (a). The airway structure is severely compromised, with luminal epithelial cells fragmentation (b, H&E), loss of ciliated epithelium, and aphazard distribution of DN-p63+ basal cells (c). Nodular/elissoid alpha-SMA+ interstitial myofibroblast foci are common, occurring in segments of severe structural changes of the airway epithelium (d). These foci, previously described and named “Sandwich Foci” (SF) [Chilosi 2006;2013], are three-layered lesions formed by a CK5+ layer of basal/oid abnormal cells (e) interposed between superficial bronchiolar epithelial cells (often non-ciliated as evidence of bnormal differentiation) and the aSMA+ clusters of myofibroblasts (c). The basal/oid cells’ phenotype include the expression of molecules related to migratory activity as laminin-5-gamma-2 (f) and heath-shock protein-27 (g,h) as well as cell senescence (p16, i). These SF, that are fairly specific for the UIP pattern, may recapitulate major pathogenic mechanisms occurring in micro-Honeycomb cyst formation, and may also represent useful “biomarkers” in small cryobiopsy samples.
These intriguing basal-type cells likely correspond to the abnormal DN-p63+ cells that have been previously recognised in bronchiolar proliferative lesions within honeycomb cysts and are characterised by unusual phenotypes (CK5-/+, CK17+, CK14+, SOX2-), expression of Wnt-pathway target genes (nuclear beta-catenin, matrix metalloproteinase 7, cyclin-D1, c-KIT), and migratory and EMT markers such as laminin-5-gamma-2 and heat-shock protein-27, tubulin-beta-3, and ZEB1.54,67,125,126 Because of their mesenchymal-like migratory phenotype, these basal senescent progenitors may progressively colonise the pulmonary parenchyma, thereby extending the remodelled nonfunctional areas and causing bronchiolisation and proximalisation of distal areas.106,108,117,125,126,127,128,129 The number of genes that have been identified to be associated with disease susceptibility has been progressively increasing, and definition of their precise pathogenic roles may facilitate the diagnosis, prognostication, and treatment of IPF. These genes include MUC5B and TOLLIP,130–132 as well as new entries such as CSK6 (proprotein convertase subtilisin/kexin type 6),133 the autophagy-related gene DEPTOR, and the mitotic spindle assembly genes KIF15 and MAD1L1.134
Clinical/pathological message: Fibrosis is a potentially misleading term because it includes a variety of lung disorders ranging from those in which cicatrisation is the pivotal process to those in which scarring is only an epiphenomenum. HC is in fact a dysplastic proliferation of aberrant bronchiolar stem cells.
Pathologic-radiologic correlations and pathogenesisThe 2018 guidelines for the diagnosis of IPF classify computed tomography (CT) findings into four subtypes: UIP pattern, probable UIP pattern, indeterminate for UIP pattern, and alternative diagnosis.135 The radiological UIP pattern, a hallmark of IPF that can also be present in conditions such as fibrotic HP, CTD-UIP, and exposure-related ILDs, is characterised by the presence of honeycombing associated with traction bronchiectasis and bronchiolectasis, mostly in the periphery of the lungs. It has a typical heterogeneous distribution with subpleural and basal predominance.135
The UIP pattern represents the final step of the ageing process following maladaptive repair that induces alterations in both the airway cellular composition and function. Therefore, if the healthy distal airway epithelium is composed primarily of mucus-producing and multiciliated cell populations, patients with IPF show misexpression of mucus and aberrant ciliation. Consequently, these patients show increased wall thickness and architectural distortion of the distal airways on multidetector CT scans, which can be more precisely visualised on micro-CT images. Micro-CT scans of the explanted lungs of patients with IPF show dramatic loss of the terminal bronchioles and a significant decrease in the alveolar surface.136 Repeated aberrant attempts at regeneration of the terminal airways through the activation of developmental pathways result in honeycomb cysts.113 The stereological analysis of HC has confirmed its spatial relationship with small conducting airways. The corresponding multidetector CT findings include an increase in the number and degree of distorted airways in the 14th to 17th generation.137 Another macroscopic consequence of this metaplastic lining process the progressive bronchiolisation of the periphery of the lungs, with traction bronchiectasis beginning to appear beneath the pleura and, over the course of the remodelling, tending to assume the aspect of HC over a continuum of aberrant lung remodelling.138 Moreover due to the large and scarcely flexible space incapable of gas exchange, another immediate consequence is the loss of elastic recoil and the collapse of HC during the maximal expiration, which has been documented by CT expiratory scans (Fig. 5).
 and espiratory CT scan (b,d) shows UIP pattern characterized by the presence of honeycombing in the anterior segment of the left upper lobe, traction bronchiectasis and focal fibrotic ground glass in the left lower lobe. A mild peripheral reticulation is also present in the right lung parenchyma. The expiratory scan (b,c) shows a homogenous reduction in lung volumes associated with a diffuse increase of the lung density, in absence of air trapping. Interestingly, the area of honeycombing doesn")
Inspiratory (a,c) and espiratory CT scan (b,d) shows UIP pattern characterized by the presence of honeycombing in the anterior segment of the left upper lobe, traction bronchiectasis and focal fibrotic ground glass in the left lower lobe. A mild peripheral reticulation is also present in the right lung parenchyma. The expiratory scan (b,c) shows a homogenous reduction in lung volumes associated with a diffuse increase of the lung density, in absence of air trapping. Interestingly, the area of honeycombing doesn't collapes (red ellipse). Moreover, the focal ground glass surrounding traction bronchiectasis, shows a lack of traction bronchiectasis collapse (red arrow).
Patients with IPF or fibrosing progressive ILD are usually diagnosed at a relatively advanced stage of the disease; however, earlier identification may allow early initiation of treatment and reduce the disease progression. Therefore, interstitial lung abnormalities (ILA) may be clinically relevant when they represent an early stage of IPF or another s fibrotic process.113,139,122 In this context, ILA, preclinical ILD, and IPF-like disorders share genetic features and a similar ageing-related pathogenetic disease profile, particularly the promoter polymorphism (rs35705950) in MUC5B, the gene encoding mucin 5B123 Hunninghake et al. found that the same MUC5B promoter variant rs35705950 increases the odds of ILA 2.8-fold (95 % CI 2.0–3.9; p < 0.001).
Evans et al. postulated that excessive production of MUC5B by stem cells that attempt to regenerate injured bronchiolar and alveolar epithelia may disrupt normal developmental pathways and enhance normal reparative mechanisms in the distal lung.113 This was recently demonstrated by histological, CT, and micro-CT analyses of eight explanted lungs or lobes with ILAs from six donors.124 The authors drew a correlation between the findings of ex vivo CT scans and the histological samples, reporting scarcely affected or near-to-normal portions of parenchyma with paraseptal fibrosis in most cases (78 %) and lymphocytic inflammation (86 %). Of particular interest is the concept of ‘paraseptal’ fibrosis, which has been described to originate from the periphery of the secondary lobule and moving inward to the centrolobule. This has already been described in surgical specimens by Colby et al. and, more recently, by Johkoh et al.140,141 The early changes in UIP patterns are characterised by an admixture of dilatation of the terminal airway and periacinar fibrosis. The peripheral acinar distribution is typical of the less fibrotic areas and can be discerned as emanating from the septa and the pleura around the bronchovascular structures.140 The corresponding imaging aspect is the reticulation, which assumes an ‘arciform aspect’ when it is subpleural or is polygonal shaped when it is far from the pleura (Fig. 6). This periacinar pattern may be identifiable in transbronchial cryobiopsy samples (Fig. 7a,b). The periacinar distribution may still be discernible on follow-up CT scans even when a typical UIP pattern appears (Fig. 7c,d). Verleden et al. also found that the opacities in or near the interlobular septa are thicker and, in more advanced stages, associated with aberrant airway-like structures that gradually fill the entire secondary lobule with progressive loss of alveolar epithelium.124 A UIP-like pattern has been described in association with pleuroparenchymal fibroelastosis (PPFE), which shows a clearly worse prognosis in comparison with other phenotypes of PPFE.142,128 While elastin fibres in the normal lung contribute to normal lung compliance and elastic return, in ILDs, overexpression of elastin leads to scarring and impaired lung function.129 An increased elastin burden has been documented in the proliferative phase of DAD and in UP as well, suggesting that it may contribute to the alveolar mechanical dysfunction and remodelling both in acute and chronic ILDs.130 In the UIP pattern, the elastin deposition is observed along the interalveolar and alveolar septal wall, and its degree correlates with the disease progression and 5-year survival.131 PPFE is also one of the most representative findings in telomere diseases, being part of a constellation of atypical or discordant findings on pathological and radiological assessments.132 The future horizons of imaging are represented by a further improvements in the resolution and the identification of coarseness. Ultra-high-resolution photon-counting CT allows more precise depiction of the lung parenchyma, with a sharper delineation of the subtle features of nonfibrotic and fibrotic ILDs.133 Finally, the implementation of deep learning algorithms for assessment of IPF represents an emerging tool that may address several unmet needs both in the research setting and in clinical practice.134,143
 shows reticulation associated with mild architectura lobular distortion, visible in the peripheral lung. This finding assumes an «arciform aspect» beneath the pleura (yellow arrow) and a poligogonal shape in the inner parenchyma (yellow circle). Scattered pulmonary ossifications are also present bilaterally. Cryobiopsy (c): the interlobular septum (star) and the centrilobular zone (arrow) are identifiable. A tongue of fibrotic tissue with an arc-like shape connects these two anatomic zones (H&E, low power).")
CT scan (a,b) shows reticulation associated with mild architectura lobular distortion, visible in the peripheral lung. This finding assumes an «arciform aspect» beneath the pleura (yellow arrow) and a poligogonal shape in the inner parenchyma (yellow circle). Scattered pulmonary ossifications are also present bilaterally. Cryobiopsy (c): the interlobular septum (star) and the centrilobular zone (arrow) are identifiable. A tongue of fibrotic tissue with an arc-like shape connects these two anatomic zones (H&E, low power).
 and twelve years later (b,c) in a 70 year old male, with history of polymiositis and dermatomyositis. In the first CT, scattered nodular ground glass opacities are present in both lower lobes (a). Moreover, some linear opacities with an arciform aspect are visible beneath the pleura, mainly in the left lobe (a, yellow circle; b, red arrow). After twelve years, the patient showed UIP patern characterized by ttraction bronchiectasis and honeycombing along the arciform lesion of the prior exam (c, yellow circle; d, red circle).")
CT scan at baseline (a,b) and twelve years later (b,c) in a 70 year old male, with history of polymiositis and dermatomyositis. In the first CT, scattered nodular ground glass opacities are present in both lower lobes (a). Moreover, some linear opacities with an arciform aspect are visible beneath the pleura, mainly in the left lobe (a, yellow circle; b, red arrow). After twelve years, the patient showed UIP patern characterized by ttraction bronchiectasis and honeycombing along the arciform lesion of the prior exam (c, yellow circle; d, red circle).
Clinical/radiological message: CT scan arciform subpleural lines may represent the periacinar distribution in UIP.
Progressive pulmonary fibrosisThe term progressive pulmonary fibrosis has been suggested to characterise a peculiar clinical phenotype (comparable prognosis and survival in IPF and non-IPF disorders).135 This concept is time-dependent. However, from a clinical point of view, the prognostication and choice of the most efficacious treatment should be performed at the time of diagnosis. The disorders grouped under this term usually have a UIP-like histopathologic background and/or share alveolar stem cell exhaustion/fibrosis/bronchiolar proliferation as a common pathogenetic trait, including the occurrence of senescent cells sharing genetic predisposition, similar molecular traits, and the occurrence of pro-fibrotic senescent ‘basaloid cells’.18,105,132,144-158,159 Therefore, this pathogenetic categorisation may be a determining step in avoiding a time-related approach.5,6,160
Clinical/pathological message: The identification of a common pathogenetic process starting from alveolar stem cell exhaustion and ending in fibrosis/bronchiolar dysplastic proliferation should be the prelude to discharge the so-called progressive pulmonary phenotype. Identification of the exact pathogenesis can eliminate the need for time to interpret the clinical behaviour of these disorders.
ConclusionsIPF and progressive ILDs characterised by the UIP-like pattern may be considered a unique category, in line with the previous thinking (‘UIP is UIP regardless of its association’).158,161 New diagnostic approaches for this disease category should incorporate genetic analysis, classical and more sophisticated imaging approaches (even those utilising artificial intelligence algorithms), histological data, as well as biomarker and immunophenotype analyses on cryobiopsies, as defined in this review.40,96,115,162,163 Validation of this scheme may provide the basis for extension of antifibrotic and new therapeutic approaches.90,164–169 In the light of this new scenario, a more precise and updated definition of the UIP pattern should be provided by integrating classical diagnostic criteria with newly acquired pathogenesis data and technical acquisitions in both imaging and pathology.40,170–172 Some issues regarding the specificity and reproducibility of classical histological criteria for diagnosing the UIP pattern have been highlighted, especially those related to the tissue samples obtained by less invasive cryobiopsies.135 A number of immunohistochemical markers that reveal subtle pathogenic mechanisms may be utilised in difficult cases.159,173–191 A summary of these criteria is provided in Table 2.
Morphological and immunohistochemical features consisting with alveolar stem cell exhaustion / fibrosis / bronchiolar proliferation (senescence driven process).
| Classical UIP pattern features170,-172 | 1. Patchy dense fibrosis with architectural distortion (i.e., destructive scarring and/or honeycombing)2. Predilection for subpleural and paraseptal parenchyma3. Fibroblast foci4. Absence of features suggestive of an alternative diagnosis (diagnostic hypothesis IPF) or ancillary findings suggesting an alternative diagnosis (lymphoid follicles, granulomas/giant cells/ peribronchiolar fibrosis, pleuritis) |
| Ancillary histological and immune-phenotypic markers173 | 1. Senescent and/or phenotypically aberrant AECII (focal DAD-like p16, p21 AECII in dense fibrosis)2.Micro-Honeycombing:-small airways'morpho-phenotypical abnormalities (basal/oid cells markers CK5/6, DNp63, CK14, MUC5B+ mucin debris)-basal/oid cells with abnormal phenotype: senescence, EMT, migratory markers, WNT activation (p16, p21; EMT: ZEB1, TBB3, nuclear beta-Catenin, MMP/, Cyclin-D1, c-Kit, Laminin-5-gamma-2, HSP27)-sandwich foci: CK5/6, Laminin-5-gamma-2, HSP27 |


