







Alpha-1-antitrypsin deficiency (AATD) is a genetic autosomal codominant disorder caused by mutations in SERPINA1 gene. It is one of the most prevalent genetic disorders, although it remains underdiagnosed. Whereas at international level there are several areas of consensus on this disorder, in Portugal, inter-hospital heterogeneity in clinical practice and resources available have been adding difficulties in reaching a diagnosis and in making therapeutic decisions in this group of patients. This raised a need to draft a document expressing a national consensus for AATD. To this end, a group of experts in this field was created within the Portuguese Pulmonology Society - Study group on AATD, in order to elaborate the current manuscript. The authors reviewed the existing literature and provide here general guidance and extensive recommendations for the diagnosis and management of AATD that can be adopted by Portuguese clinicians from different areas of Medicine.
This article is part of a supplement entitled “Portuguese consensus document for the management of alpha-1-antitrypsin deficiency” which is sponsored by Sociedade Portuguesa de Pneumologia.
Alpha-1-antitrypsin (AAT) – the major serine protease inhibitor in the serum – is primarily synthesized by hepatocytes and also produced at lower concentrations by neutrophils, mononuclear phagocytes, and epithelial lung and intestine cells. Once secreted into the bloodstream, AAT weighs approximately 52 KDa, is composed of 394 amino acids and three carbohydrate side chains linked to asparagine residues.1,2 Structurally, this globular glycoprotein presents three β-sheets (A-C), nine α-helices (A-I) and a solvent exposed stretch named reactive centre loop (RCL). There, the peptide bond Met358-Ser359 (P1-P1’) mimics a protease substrate and, upon its cleavage, AAT initiates a major conformation rearrangement that culminates in the formation of a stable inhibitor-protease complex.3–5 Although AAT is well established as a potent inhibitor of neutrophil elastase in the lung, it is also capable of inhibiting other proteinases, such as proteinase 3, kallikreins 7, matriptase and caspase-3. More recently, immunity-related and anti-inflammatory functions have also been attributed to AAT.1,6
AAT is encoded by the SERPINA1 gene located in the chromosome 14q32.1 region. This gene encompasses approximately 14kb and is organized into three untranslated exons (Ia-Ic) containing different tissue-specific transcription start sites and four coding exons (II-V).7
AAT deficiency (AATD) is inherited as an autosomal codominant condition, which is caused by pathogenic mutations in SERPINA1. There are currently over 120 identified alleles that follow a specific coding system, in which inherited AAT variants are classified between ‘A’ and ‘Z’ based on their migration in an isoelectric focusing (IEF) pH gradient (‘A’ denotes the most anodal variants and ‘Z’ the slowest migrating alleles – see also section 3.2.3.1).
AAT variants can be categorized into four groups8:
- -
Normal variants, characterized by normal AAT serum levels (20–53μM, or 80–220mg/dl by nephelometry).
- -
Deficiency variants, characterized by AAT serum levels below 20μM, and, in the case of some alleles (i.e. Z), concomitant decreased functional activity of the AAT molecule.
- -
Null variants (Q0), which are characterized by the absence of circulating AAT due to either transcriptional or translational errors that prevent normal protein synthesis.
- -
Dysfunctional variants, which are characterized by an abnormal function of AAT (i.e. reduced binding to neutrophil elastase, which is the case of F and Z variants, or with altered inhibitory activity, which is the case of anti-thrombin Pittsburgh variant).9
Normal alleles are present in 85–90% of individuals and are designated as M; therefore, normal individuals reveal MM genotypes. On the other hand, most prevalent deficiency alleles are designated as S and Z.10 The Z allele results from a Glu342Lys substitution that causes the loss of a crucial salt bridge (Glu342-Lys290) and affects the stability of the A β-sheet (breach and shutter domains).3 This structural change is thought to facilitate the insertion of the RCL into the shutter of another AAT molecule prompting the accumulation of Z polymers in the endoplasmic reticulum (ER).2 The S allele is caused by a Glu264Val replacement that also leads to the disruption of a salt bridge (Glu264-Lys387) linking an α-helix to the molecule core.3 The S variant is also prone to polymerization in the ER; however, its rate is only marginal when compared to the Z allele.1,11,12
EPIDEMIOLOGYEpidemiological studies show that AATD is one of the most prevalent genetic disorders in humans and the most commonly diagnosed in adults. Over the past 10-15 years, diagnosis of AATD has markedly improved as a result of increasing awareness and the publication of international diagnostic and management evidence-based recommendations. Nevertheless, this condition remains widely and persistently under-diagnosed or misdiagnosed by healthcare providers. Even in Sweden, where AATD was first recognized and reported, Piitulainen and Tanash stated recently that only 20% of all expected cases of severe AATD in adults were reported to the Swedish national registry.13
Despite scarce epidemiological information on a worldwide basis, several studies showed that AATD is relatively common among Europeans and its frequency can be compared to that of cystic fibrosis, which affects one in every 2000 to 4000 people.14 Nonetheless, the study carried out by de Serres15 – which included 58 countries worldwide and a total population of approximately 4.4 billion inhabitants – obtained an estimate of at least 116 million carrying AATD MS or MZ and approximately 3.4 million deficiency allele combinations (SS, SZ, and ZZ). This study also concluded that AATD is an inherited condition affecting all racial subgroups, not just Europeans but rather several African populations, middle eastern Jews and Arabs, and Asians as well. Blanco et al16 carried out a multicentric study involving 21 European countries in order to estimate the number of carriers for the two most common deficiency alleles (S and Z). This study revealed that Italy, Spain, Germany, France, United Kingdom, Latvia, Sweden, and Denmark have the highest number of ZZ carriers (5000-15000) followed by Belgium, Portugal, Serbia and Montenegro, Russia, Holland, Norway, and Austria, the latter of which have between 1000 and 2000 carriers.
Over 90% of individuals with clinical deficiency carry the ZZ genotype, while the remaining ones are explained by other rarer variants.17 The Z allele is most common in northern Europe, in those of Scandinavian descent, nearly absent among Africans and Eastern populations, except where there is miscegenation with Europeans, while the S allele has increased in the Iberian Peninsula. AATD is always associated with non-Z alleles in Asian populations. The null allele occurs in a frequency of 0.00017.14,17
Few epidemiological studies have been carried out in order to quantify the true prevalence of this genetic deficiency and information on the Portuguese population remains scarce. One study described the geographic distribution of alleles across Portugal with S allele having a decreasing frequency from north to south and Z allele reaching maximum frequency in the north and minimum frequency in the centre.18 The most relevant data published refers to a retrospective study that evaluated a sample of the Portuguese population tested for AATD over 10 years.19 Data from 1684 patients from almost every region in Portugal – whose blood/plasma samples were sent to an informal reference laboratory – were analysed. It was possible to identify 427 MZ, 188 SZ, and 158 ZZ subjects. Different types of rare deficiency and null alleles were also detected: 59MMalton or MPalermo (p.Phe52del), 28 Q0Ourém (p.Leu353Phefs*24), 21 PLowell (p.Asp256Val), 17 I (p.Arg39Cys), 6MWürzburg (p.Pro369Ser), 6MHeerlen (p.Pro369Leu), 4 T (p.Glu264Val), 2 Q0Madrid, 2 Q0Faro (c.-5+2dupT), 2 ZAugsburg (p.Glu342Lys), and 1 Q0Lisbon (p.Thr68Ile) variants. Four alleles linked to novel mutations were also identified: PGaia (p.Glu162Gly), Q0OliveiradoDouro (p.Arg281Lysfs*17), Q0VilaReal (p.Met374Leufs*19), and SGaia (p.Leu118Phe).20,21
DIAGNOSIS OF ALPHA-1 ANTITRYPSIN DEFICIENCYIn 1963, Carl-Bertil Laurell and Sten Eriksson first described AATD as a clinical entity.22 Since then, there have been impressive strides in understanding the pathobiology and clinical course of this inherited disorder.23 Nevertheless, many studies continue to identify AATD as an under-recognized condition in symptomatic patients. A five to eight-year delay between the onset of the first symptom (usually dyspnea) and the recognition of AATD has been found in studies performed over a time span of 18 years up till 2013, and there has been no overall improvement in early disease detection.24–27
CLINICAL MANIFESTATIONSAATD by itself is not a disease, but rather a predisposition to a later development of several disorders. In fact, it is believed that low serum levels of AAT together with other genetically determined characteristics and environmental influences result in the development of a disease state that may result in premature death. The available data on penetration (the percentage of AATD individuals with clinical disease) is currently scant and the spectrum of AATD-related disorders and their ages of onset are quite broad.28
In clinical practice, the risk of disease is mainly restricted to the deficient Z allele, accounting for approximately 95% of AATD cases. The remaining individuals at risk carry rare alleles including null and dysfunctional variants.8
Low levels of AAT represent a syndrome of clinical entities, some more related to a deficiency condition while others resulting, in contrast, from a negative effect of protein overload. Its main clinical manifestations primarily involve the lungs, liver, and – less frequently – the skin. Since patients with AATD are susceptible to developing all these diseases throughout their lives, the disorder is considered a systemic disease.29
Individuals may be identified after presenting clinical consequences of the deficiency or through family screening of an index case.13,30
Currently, the understanding of the natural history of AATD is still incomplete. Within the first three decades of life, liver disease is the major threat and pulmonary dysfunction is not common. Besides that, the natural history of AATD patients is less clear, and survival estimates vary widely among series.
1.1PULMONARY INVOLVEMENTIn the lung, AATD predisposes individuals to premature onset of chronic obstructive pulmonary disease (COPD). Indeed, it is the major and best proven genetic risk factor for COPD, with 1-2% of all cases estimated to be due to severe AATD.28,31
1.1.1EmphysemaThe premature onset of emphysema was the first identified clinical manifestation of AATD, described in 1963,22 and it is the most prevalent clinical correlate of AATD and the major cause of morbidity and mortality.32 Emphysema is thought to primarily result from a ‘loss of function’ defect.8 It corresponds to a protease-antiprotease imbalance and proteolytic degradation of elastin and other extracellular matrix components of the respiratory tract by neutrophil elastase (NE) and other proteases, the activity of which is unopposed and enhanced because of AATD in the pulmonary parenchyma and airways. Aside from a quantitative deficiency in the serum, Z-type molecules in their monomeric isoforms are approximately fivefold less efficient inhibitors of NE than normal native M-type AAT.33,34
Compounding the lack of a proteolytic screen, the transition of monomeric Z-AAT to polymers inactivates its anti-proteinase function, thereby further depleting protective levels of AAT, and also converts this natural broad-spectrum anti-inflammatory molecule into a proinflammatory stimulus. Loop-sheet polymers of misfolded Z-types have been shown to cause chemotaxis and/or to activate inflammatory cells, hence supporting the hypothesis that polymers in the lung (either circulating and trapped in the lung or locally produced by alveolar macrophages and airway epithelial cells) may contribute to tissue destruction.35 Moreover, the accumulation of misfolded AAT molecules within the ER lumen of mononuclear phagocytes and neutrophils leads to ER stress response that may also have a role in the pathophysiology of lung disease.36 Indeed, the presence of polymers may explain the progression of lung disease in Z homozygotes after smoking cessation despite appropriate intravenous replacement with plasma AAT.37
Individuals with genotypes associated with plasma AAT levels below the putative protective threshold of 11μM (corresponding to 57mg/dl by nephelometry or 80mg/dl by immunodiffusion), most of all homozygous for the deficient Z allele, are considered to have severe AATD.8 Consequently, they are at a markedly increased risk of early development of panacinar emphysema,28 particularly when exposed to cigarette smoke, environmental irritants, or infections.25 However, the precise risk of developing emphysema in individuals with severe AATD is not completely understood.
Although several studies suggest a high risk of emphysema in ZZ subjects, some never develop lung disease. Accordingly, a post-mortem series from Sweden38 that included computed tomography (CT) imaging studies39 showed that 14 to 20% of Z homozygotes can be free of emphysema.
Individuals with null alleles are at risk of the most severe form of associated lung disease, including emphysema.
Although S homozygotes are not associated with a significant risk of emphysema, SZ heterozygotes have an increased risk, particularly if associated with cigarette smoking.40–43 Moreover, several studies performed so far, with differences in terms of sample sizing, have provided contradictory results about an increased risk of COPD in MZ individuals.31,44–47 However, more recent studies based in large populations of patients and controls support an increased risk of COPD mainly influenced by exposure to cigarette smoke.48
Findings from the National Heart, Lung and Blood Institute Registry (NHLBI) show that individuals with severe AATD display the same symptoms as patients with ordinary, or non-AATD, COPD. These symptoms (in order of decreasing frequency) include dyspnea on exertion (84%), wheezing with upper respiratory tract infections (URTI) (76%), wheezing without URTI (65%), increased cough and phlegm (50%) and chronic cough (42%).30
Holm analyzed cross-sectional data collected via self-report questionnaires completed by 480 individuals with non-AATD COPD and 578 individuals with AATD-associated COPD. Individuals with AATD-associated COPD did not report more symptoms of depression (25% reported depressive symptoms in the clinical range) or anxiety (36% reported clinically relevant symptoms of anxiety) despite their younger age; however, they did report more clinically significant dyspnea and greater impairment to health-related quality of life (HRQoL) than other individuals with COPD.49 In addition, a study which assessed the quality of life of Portuguese patients with AATD showed that female patients had worse HRQoL. Hospitalizations and functional markers of respiratory disease progression negatively influenced the HRQoL.50
Considerable variability in the time of symptom onset has been described, but symptoms rarely appear before 25 years of age. Severe symptoms are most often seen in current or former smokers but some smokers and many non-smokers develop no symptoms at all.27 Cigarette smoking can accelerate the onset of dyspnea by as much as 22 years. In three studies the age at onset of dyspnea was 48 to 54 years in non-smokers and 32 to 40 in smokers.32,51,52
Distinctive features of emphysema associated with severe AATD are early onset (i.e. age ≤45), being present in a non-smoker or minimal smoker who has no occupational risk factors and basilar-predominant radiographic changes.28 Individuals with severe AATD experience a wide variation in lung function, usually with an accelerated decline. Estimates for the annual rate of decline of forced expiratory volume in one second (FEV1) in Z homozygotes vary between 23 and 316ml and the rate of decline is strongly affected by cigarette smoking.8
The onset of airflow limitation typically occurs at a younger age than in usual for COPD. In a multicenter natural history study of Alpha1-Antitrypsin Deficiency Registry Study Group, the mean FEV1 of the 1129 participants was 43±30% of that predicted and their average age was 46±11 years.27 In non-AATD COPD patients, airflow limitation usually appears in the sixth and seven decades of life.
Several studies have shown FEV1 values to be a major determinant of survival in severe AATD. Two year survival is practically 100% until FEV1 falls to 33% of that predicted and, from this point on, it decreases exponentially, falling to 50% when FEV1 is 15% of that predicted.53
With COPD, airflow obstruction usually coexists with a reduced capacity of carbon monoxide diffusion; however, discordance is not infrequent. Even patients with severe airways obstruction and prominent panacinar emphysema on the CT scan may have normal gas transfer.54
Variations in the distribution of emphysema may be associated with functional differences and therefore account for discordant physiology. Basal distribution of emphysema has been associated with greater impairment of FEV1 but lower impairment of gas exchange than the apical distribution. It is possible that real physiological differences do exist between the upper and lower lung regions, and these may be responsible for the functional differences identified.55
The wide range of clinical phenotypes that are associated with AATD could be caused by interactions between genetic modifiers and environmental factors other than SERPINA1 gene cluster and just smoking.36
The detrimental role of exposure to cigarette smoke on the clinical phenotype of AATD has been widely demonstrated, with a significant gene-by-smoking interaction. Smoke exposure is the single greatest determinant for the progression of emphysema in AATD and a key element in the natural history of those patients.56 There appears to be a dose-response relationship between cigarette consumption and change in FEV1 over time.57 Passive smoking may also be a risk factor for respiratory symptoms, but no relationship was found between second-hand smoking and decreased lung function.58
Active smoking – often but not always associated with lower FEV1 – does not fully explain the variability, and there are still variations in the clinical course of patients who have never smoked.59 The natural history of AATD in never-smokers is altered at some point in symptomatic individuals, by an unknown trigger, worsening their condition and bringing them to medical attention.56 Other risk factors for airflow obstruction may be male gender, a parental history of COPD, and a personal history of asthma, chronic bronchitis, or pneumonia, among others.60,61 The precise role of occupational inhalation exposure in accelerating the loss of lung function in AATD patients is not completely understood.62,63 There are few data available to indicate that long-term outdoor air pollution (ozone and primary particles) can worsen the respiratory status and predict lung function decline in individuals with the ZZ genotype.64,65
In addition to smoking and environmental exposure, factors associated with faster decline of lung function in AATD may include male gender, age 30 to 44 years, low body mass index, lower serum AAT level, exacerbation rate, bronchodilator reversibility, and baseline severity functional capacity.66–68
1.1.2COPD exacerbationsAATD exacerbations are associated with greater inflammation and elastase activity than is usual with COPD, which may explain why exacerbations can be more frequent, prolonged, and severe.69 These episodes may relate to physiological decline even in patients undergoing augmentation therapy.70 In a large one year cohort study from United Kingdom, neither the presence nor the frequency of exacerbations showed a relationship to the decline in FEV1, but the number of exacerbations was weakly associated with a decline in lung gas transfer for carbon monoxide.71
1.1.3Spontaneous secondary pneumothoraxThis acute disease may be the presenting manifestation of AATD72 or a complication of emphysema.73 To date, the mechanisms of spontaneous pneumothorax are still unclear. Preliminary data of a case control study of 39 patients with the diagnosis of spontaneous pneumothorax confirm the clinical importance of AATD phenotypes in those patients and the need for AATD screening.74
1.1.4AsthmaThere is an unclear relationship between AATD and asthma. Diagnostic bias and the overlap of COPD with asthma may be a confounding factor in the evaluation of asthma prevalence among AATD subjects. Although several studies failed to prove an increased prevalence of AATD among asthmatic patients,75 asthma has been found to be the most common respiratory complaint in patients with AATD prior to the disease diagnosis.27
A review published in 2010 summarizes information on the relationship between severe AATD, asthma, and COPD.75 Current literature indicates that asthma signs and symptoms are common and may occur early in the course of the development of airflow obstruction in AAT-deficient patients with or without COPD. Bronchodilator response is a risk factor for FEV1 decline and AATD itself predisposes to airway hyperresponsiveness that is associated with reversible airflow obstruction. The difficulty in differentiating asthma and COPD in those patients is particularly challenging since wheezing and airflow obstruction are common manifestations of both conditions. An increased awareness by clinicians of the overlap between these two conditions is fundamental to ensure appropriate therapeutic strategies in order to prevent an accelerated decline of lung function.75
1.1.5BronchiectasisBronchiectasis has also been associated with severe AATD, but the relationship between these conditions remains unclear and the estimated prevalence varies widely across different reports.41,76,77
The Portuguese recommendations are for the aetiological diagnosis of bronchiectasis; given the high frequency of deficient alleles in the Portuguese population and the benefit of early AATD detection, testing for AATD should be undertaken for all subjects with bronchiectasis.78
EXTRAPULMONARY DISEASEIn addition to extrapulmonary disease, liver and skin disorders may also occur in AATD. Clinical evidence of an association with other diseases such as bowel disease, glomerulonephritis, rheumatoid arthritis, fibromyalgia, vascular abnormalities (fibromuscular dysplasia of the arteries, abdominal and brain aneurysms, and arterial dissection), psoriasis, chronic urticaria, pancreatitis, neoplasia, and multiple sclerosis, is rare and further research studies are required prior to reaching any conclusions.79
Liver DiseaseLiver disease does not occur frequently. AATD is only diagnosed through finding of liver disease in 3% of AATD cases.80 Unlike pulmonary disease, AATD-related hepatopathy is not caused by functional loss or low protein serum levels but rather by the accumulation of AAT polymers in hepatocytes.81 In this context, liver disease is the result of a disrupted balance between the accumulation of AAT polymers and the capacity of cellular mechanism to degrade those protein aggregates.81,82 Hence, those AATD variants linked to polymerization in the liver, such as SIiyama, MDuarte, MMalton, and particularly Z allele, may all show signs of hepatopathy. Liver disease has never been reported in Null (Q0Q0) patients. Unlike pulmonary disease, which is typically a disorder of adulthood, liver disease may occur throughout a patient's life; it is a frequent cause of disease in non-smokers. The prevalence of post-mortem liver disease in non-smokers is 28%. Approximately 10-15% of newborns with severe AATD are at risk of developing some form of liver disease. Similarly, about 10 to 15% of adults show some evidence of liver disease, and although it may occur at any given age, it occurs more frequently later in life.80 Cholestatic jaundice and hepatitis (neonatal hepatitis syndrome) can be observed in some new-borns with AATD, while later during childhood AATD patients may experience an unexplained increase in aminotransferases, hepatomegaly, or on rare occasions, cirrhosis and liver failure. In some cases, the only signs of liver disease in young children may be anorexia and a growth delay.83 On the other hand, adults may show asymptomatic changes in transaminases, clinical signs of cirrhosis, and hepatocellular carcinoma. Clinical manifestations in patients with chronic liver disease due to AATD may not be differentiated from those of any other etiology.
The development of liver disease and its evolution towards cirrhosis is the most common clinical sign after pulmonary disease. It is estimated to occur in 2-43% of ZZ patients. It may be a frequent cause of death in non-smokers with the ZZ phenotype.84 According to a Swedish study, out of a population of 200 000 newborns, 22/120 Z individuals (18%) showed signs of some liver dysfunction, which included obstructive jaundice (12%) and discrete lab changes (7%).85 The risk of cirrhosis in these cases was approximately 50%, approximately 25% died during the first decade, and 2% developed cirrhosis later on in their childhood.86 The follow-up of 127 ZZ newborns until their 30s showed that 3-7% had high transaminase levels, but none of them showed any clinical symptoms of liver disease.87 In a second study where 246 ZZ patients were followed over an 11-year period, Larsson observed liver disease in 12% of the cases (cirrhosis in 11.8%, neonatal hepatitis in 0.4%, and hepatoma in 3.3%).32 ZZ individuals who develop cirrhosis are at an increased risk of developing hepatocellular carcinoma,88 especially middle-aged or older men, although there have been some rare cases described in children.89 However, this risk has been difficult to quantify and may vary across studies.88
The risk of heterozygous patients developing liver disease is not well established. Many studies have shown that there is a greater prevalence of the MZ genotype among patients with cirrhosis than in control populations with non-cirrhosis liver disease or cryptogenic cirrhosis.90–94 However, a more recent study has not shown any association between the MZ genotype and liver disease.95 The risk of individuals with one Z allele developing hepatocellular carcinoma is also not as well defined as for homozygous patients. Heterozygosity for the Z allele may increase the risk or change it in patients with other liver diseases, such as chronic hepatitis C.96
PanniculitisPanniculitis was described for the very first time by Warter in 1972 and it has been associated with AATD in approximately 50 reported cases. Panniculitis is very rare with an estimated prevalence of 1 per 1000 AATD cases,30 and with an average age of appearance of 40 years. As with emphysema, panniculitis is thought to occur due to a lack of protease inhibitory activity in the skin. It is characterized by pain and one or more nodules showing exuding liquid and redness, and painful and warm cutaneous plaques, which may suffer necrosis and be recurrent. Although it occurs more often on the thighs and buttocks, in one third of cases it will occur in places of trauma. There are rare situations where it may be accompanied by polyserositis. It may occur in several disease genotypes, including ZZ, SZ, SS, and MS.97 Diagnosis requires deep excisional biopsy, which shows areas of fat necrosis interspersed among normal areas. A differential diagnosis with other causes of panniculitis is difficult and that is why AAT levels should be measured in every case of necrotising panniculitis.
VasculitisAn association between antiproteinase 3 (PR3) antibody vasculitis (c-ANCA positive) and AATD was first reported in 1993.98,99 Since then, several series have established a correlation between AATD phenotypes and anti-PR3 vasculitis. However, there is a low incidence of antiproteinase 3 antibodies in patients with AATD and its presence may not be related to the development of systemic vasculitis.100 Taking into account this association, AATD testing must be carried out in adults with c-ANCA positive vasculitis.28
LABORATORY TESTSDiagnostic tests to confirm the presence of AATD are divided into quantitative and qualitative tests. There are four types of tests:
- –
Serum AAT Test, quantitative test for determining the amount of protein in the blood;
- –
AAT Phenotyping, qualitative test that assesses the protein variants in the blood. This technique is capable of detecting normal alleles (M) and deficiency alleles (S, I, Z, and others) but it will not detect null alleles (i.e. Q0Ourém);
- –
AAT Genotyping,
- -
Searching for S and Z mutations, qualitative test that assesses the presence or absence of the most common mutations in deficiency - S and Z alleles.
- -
SERPINA1 gene sequencing, qualitative test that is considered the reference method for identifying specific mutations associated with AAT rare alleles, whether they are deficient or null (I, MMalton, PLowell, Q0Ourém, etc.).
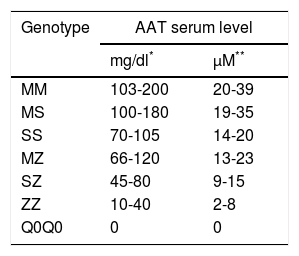
The concentrations of AAT serum levels may be measured using several different methods: immunoelectrophoresis, radial immunodiffusion and nephelometry, and normal values will vary depending on the method used101 (Table 1). Nowadays, the most commonly used method is nephelometry. Radial immunodiffusion and nephelometry tend to overestimate the concentration of AAT when compared to the purified standard test developed by the US National Institute of Health. In order to differentiate the values obtained using the non-purified standard test from those obtained using the purified standard test, the former are expressed in mg/dl while the latter are in μM. Both units are frequently used and are interchangeable in many European countries, whatever the test used. In order to convert mg/dl (nephelometry) to μM, one must multiply the concentration in mg/dl by 0.1923.79
The reference values for each method are shown in Table 1. In order to make it easier to interpret results, each laboratory must previously determine the normal values of AAT in the serum samples of normal individuals.79
Bearing in mind that different alleles determine a different percentage of protein production, there are expected serum levels for each phenotype79 (Table 2).
AAT behaves like an acute phase protein, which is why its serum levels may be falsely augmented during inflammatory and infectious processes. Therefore, AAT tests must be carried out preferably outside these episodes. Higher AAT values can be also found during pregnancy and after taking oral contraceptives. In these cases, normal serum levels may be detected in patients with moderate deficiency. Serum levels are lower in children than in adults.79
DETERMINING THE NEUTROPHIL ELASTASE INHIBITORY CAPACITYIn rare circumstances, another test may be used to assess if a patient with normal circulating levels has an AATD. The inhibition of NE by AAT is directly related to its serum concentration in such a way that a greater amount of AAT circulating will have greater inhibitory effect. However, some patients may present normal or high AAT levels and little anti-elastase activity due to the fact that not every protein is active (80% be oxidised, destroyed, or not capable of linking to neutrophils).
Measuring the capacity to inhibit elastase from neutrophils is especially indicated in the following cases: 1) studying patients with COPD where AATD is suspected although with a normal AAT serum level; 2) when determining if during an exacerbation of COPD the increase in AAT is proportional to anti-elastase activity; 3) when assessing the implication of AAT deficiency in the severity of pulmonary disease, since quite often AAT serum concentration is not related to clinical progress.79
DETERMINING PHENOTYPE AND GENOTYPEQualitative tests detect the different mutations of the SERPINA1 gene. While phenotyping is carried out using isoelectric focusing (IEF) techniques that allow for the identification of several protein variants in the blood (S, Z, M, and others), genotyping usually covers different variations to the polymerase chain reaction (PCR) method, which enables the analysis of different mutations in more or less extensive regions of the SERPINA1 gene.
1.1.6PhenotypingSeveral protein variants present in the blood, corresponding to different alleles of the SERPINA1 gene, may be identified through isoelectric focusing.
Although phenotyping permits the characterisation of approximately 30 AATD variants, this method alone will not be sufficient for a complete diagnosis in the case of null variants with very low (MHeerlen) or virtually non-detectable (Q0Ourém) protein serum levels. Phenotyping may also lead to a partial diagnosis in the case of M-like (MMalton and MWurzburg) variants which, in addition to presenting reduced concentrations of protein, show a very similar migration pattern to M protein.
1.1.7GenotypingDifferent methods based on the PCR technique have been used for genotyping the mutations that define the S (rs17580, p.Glu264Val or c.863A>T) and Z (rs28929474, p.Glu342Lys or c.1096G>A) alleles, which use either primer oligonucleotides or specific fluorescent probes (in the case of real time PCR detection), detecting exclusively the mutated (S and Z) or non-mutated (non-S and non-Z) position in the DNA sequence.102 In spite of these methods being technically simpler and more likely to be applied to a broader genetic screening of AATD, they do not enable us to detect the presence of other mutations which occur in Portugal.21 If while using these techniques S and/or Z alleles are not detected, patients will be frequently catalogued as carrying the M allele (homozygosity or heterozygosity) since in the case of the Portuguese population the most frequent alleles are M (80-86%) followed by S (12-18%) and Z (1-2.5%).103,104 This field is rapidly advancing and soon there will be diagnostic kits capable of detecting a greater number of mutations of the SERPINA1 gene.
Only when genotyping includes the amplification of SERPINA1 coding regions (exons II to V) and DNA sequencing does it become possible to assess in a systematic manner the AATD genetic variation. It may be complemented by an analysis of the exon-intron regions and the promoter (exons Ia to Ic) in order to rule out eventual mutations that may affect the normal processing of mRNA (Q0Oporto and Q0Faro).104 Although this form of genotyping may represent an excellent method for diagnosing AATD, currently its costs and technological requirements, particularly in less specialised laboratories, still prohibit its use as a screening tool. Hence, this exceptional method is only used to confirm diagnosis in the case of rare AATD alleles that have been identified through isoelectric focusing (phenotyping), in particular situations where serum levels diverge from phenotype and/or genotype (S and Z mutation search), when results are inconclusive or do not agree with the clinical manifestations, or whenever other rare or null variants are suspected.79
Considering the complexity of the AATD genetic diagnosis, partly due to the large number of rare variants,104 and the specificities and limitations of each different technique – genotyping and phenotyping –, the different reference laboratories in Europe have established different diagnostic algorithms, which often combine the measurement of AAT serum levels, the search for S and Z mutations, isoelectric focusing, and the SERPINA1 gene sequencing in order to reduce the number of false negatives and positives.105
Phenotype and/or genotype may be determined in a total blood sample preferably resorting to the ethylenediamine tetraacetic acid (EDTA) anticoagulant, which should be processed into two segments within an approximate timespan of 48h: plasma or serum (for isoelectric focusing) and mononuclear blood cells or total remaining blood (for genomic DNA extraction). In any case, the different blood fractions may be stored at -4°F (-20°C).79 The timely execution of this procedure is crucial in order to identify deficient variants, such as the Z allele, given its lability and consequent risk of deterioration.
Recently, as an alternative to collecting samples by venipuncture, rapid genetic screening methods (kits) have been provided, which enable a quick assessment of the presence of S and Z alleles in blood (Alphakit®) or saliva samples. However, these tests still require result confirmation using more conventional methods, particularly if one of these alleles is detected, or if AATD is strongly suspected. Currently, there are also AATD genetic screening programmes, which favour sample collections using minimally invasive methods by means of finger puncture and collection of a small amount of blood on dry filter paper (DBS: dried blood spots), which may be sent by post without any extra care. Several studies have shown that testing and searching for S and Z mutations by means of a dried blood drop sample is feasible and it is particularly suitable for the purpose of screening in a larger population, since it is a simpler and cheaper sampling method.105–109 However, due to the lower quantity and quality of the obtained sample, in some cases – particularly regarding the SERPINA1 gene sequencing and phenotyping – it may be necessary or even desirable to confirm results by means of a total blood sample collected by venipuncture.28,105
1.1.8AATD GenotypesGenotypes that increase the risk of developing pulmonary disease are associated with serum values lower than 11μM or 57mg/dl (protective threshold) which may result from different combinations of deficient (Z, MMalton or other rare alleles) and null (Q0Ourém or others) alleles in homozygosity or heterozygosity such as ZZ, SQ0, ZQ0, Q0Q0 or with levels of serum similar to these (Table 2). Intermediate AATD is characterised by serum concentrations between 11 and 20μM and associated to several genotype combinations in homozygosity or heterozygosity, such as SS, SZ, MZ, MS, SQ0, and MQ0, among others79,101 (Table 2).
On the other hand, genotypes that have been described as being associated with a greater risk of liver disease are those associated with homozygosity or heterozygosity for alleles with proven susceptibility to form polymers in hepatocytes (Z, S, MMalton, MPalermo, MNichinan, SIiyama and MWurzburg).2,21
DIAGNOSTIC ALGORITHMThe AATD diagnostic algorithm (Fig. 1) begins with a clinical suspicion of AATD (Table 3). In this case, the quantification of AAT serum levels is recommended by doing a quantitative test, often by nephelometry.28

Clinical cases where an AATD screening is recommended.
| Clinical Suspicion of AATD: Candidates for AAT values to be determined |
| All COPD patients |
| Early pulmonary emphysema (patients younger than 45) |
| Emphysema when not exposed to known risk factors (smoking and occupational factors) |
| Panlobular emphysema that is predominantly basal |
| Asthma with spirometry that does not normalize in spite of adequate therapy |
| Adults with bronchiectasis |
| Teenagers with a persistent obstruction in lung function tests |
| A clinical history of dyspnea and chronic coughing in several members of the family |
| Hepatopathy of unknown cause |
| Decrease in the peak of the alpha-1 protein in the proteinogram |
| Panniculitis or vasculitis of unknown cause |
| Relatives of AATD patients (brothers and sisters, offspring, parents, others) |
The consensus document from the American Thoracic Society and European Respiratory Society (ATS/ERS)28 suggests the combination of the quantitative test and phenotyping as gold standard for the diagnosis of AAT deficient states. In a recent document updating the Spanish guidelines,110 this algorithm continues to be recommended with high quality scientific evidence, since it enables us to clarify most AATD cases. However, some authors defend the association of a quantitative test with genotyping as the best method for diagnosis.105,111,112 Whatever the test used, there should be a concordance between the identified phenotype/genotype and the measured serum level (Table 2).
According to ATS/ERS28 recommendations, individuals with a AAT serum level below the normal value, or between 90-140mg/dl through nephelometry, which are considered as borderline values, should undertake a qualitative test that includes phenotyping and/or genotyping, since these values may correspond to a phenotype with intermediate deficiency (SZ, SS, MZ) and there may be relatives with a severe deficiency. According to Ferrarotti,113 the following cut-off points were proposed: 1) If the purpose is to identify genotypes with an increased risk of emphysema, then a 100mg/dL cut-off point is sufficient (95.8% sensitivity, 94.8% specificity); 2) If the purpose is to detect all patients carrying Z and S alleles, then a 110mg/dL cut-off point should be used (73.4% sensitivity, 88.5% specificity); 3) Other studies have used a 113mg/dL cut-off point, since there are no Z alleles or SS individuals above this value (100% sensitivity, 78% specificity for AATD and 79% sensitivity and 83% specificity for detecting Z and S alleles). There is a high number of false positives above these values.114
Based on this, we recommend a qualitative whenever the serum level is below 110mg/dl.
The genetic diagnosis of AATD should be supported by reduced serum levels (<110mg/dl) and a compatible phenotype or genotype that is recognised as being related to the disease. Whenever the serum levels, phenotype, or genotype do not agree with each other or with clinical manifestations, then a rare, deficient, or null variant should be considered, and it should be investigated by a reference laboratory in order to clarify the situation.
SCREENINGIn light of the misdiagnosis and delay in the diagnosis of AATD, in Portugal and in the rest of the world, it is mandatory to screen risk groups if we wish to change this panorama. Early diagnosis and proper follow-up should lead to preventive attitudes and lifestyle changes that will certainly change the progress of this disease – smoking, occupational exposure, and infections –, early treatment of symptoms and exacerbations and administration of replacement therapy in the early stages of the disease in order to improve life quality and expectancy of these patients. Therefore, it is imperative to identify the disease as soon as possible.
However, identifying individuals with AATD may also carry some risks, such as genetic discrimination, a high psychological burden, and may have employment implications. Hence, before doing the screening, doctors should weigh the pros and cons of diagnosis and discuss them with the patient.28
Screening is recommended for patients who are clinically suspected for AATD (Table 3). In the case of screening relatives of the index case, it has not yet been established how wide this screening should be, although some advise that first degree relatives should be tested.115
The screening method should be similar to that used in the diagnosis and follow the same algorithm.
The implementation of national screening programmes is leading to an increase in the number of detected cases. Countries where screening programmes have already been implemented116–119 show different detection rates. Many of these programmes are carried out as an initiative of the AATD National Registries. When designing a screening programme for detecting AATD, one must consider the sample process protocol as well as criteria for including candidates, since both factors will have a decisive impact on the programme results and cost.120 We suggest screening programmes using accurate and simple methods with dried blood drop samples where AAT is tested and phenotyping and/or genotyping is carried out using fast methods which are cost-effective.
- -
AATD diagnosis begins with a clinical suspicion that should be confirmed by AAT serum levels and phenotyping and/or genotyping.
- -
AAT serum levels <110mg/dl (nephelometry) should prompt to phenotyping and/or genotyping
- -
There should be agreement between serum levels and the phenotype/genotype, otherwise we could be in the presence of a rare allele.
- -
If there is suspicion of a rare allele, samples should be sent to a reference laboratory in order to make a correct diagnosis.
The initial assessment of a patient with AATD should begin with a medical history and physical exam with special focus on pulmonary disease as well as in the other clinical manifestations of the disease, such as liver and skin disease.
In addition to the AAT test and phenotyping/genotyping, which were carried out during screening and diagnosis, additional exams should be undertaken in the early stages of a patient with confirmed AATD (Table 4).
Additional exams in the early stages of confirmed AATD.
| Pulmonary Assessment | Liver Assessment |
| Spirometry | Liver function tests |
| Pulmonary volumes | Abdominal ultrasound (or Fibroscan TM) |
| DLCO | |
| Arterial blood gas analysis | |
| Chest X-ray | |
| Chest CT (with emphysema quantification, if available) | |
| Six-minute walk test |
The initial assessment of a patient with AATD should include: spirometry – before and after a bronchodilator –, pulmonary volumes – measured either through plethysmography or the helium dilution technique or nitrogen-washout –, carbon monoxide diffusion capacity, arterial blood gas analysis, and 6-minute walk test.28 Other respiratory function exams may be carried out in selected cases.
Spirometry is the most suitable exam for the diagnosis of COPD and for assessing its progression. There is usually an obstructive pattern (FEV1/FVC<0,7) with a decrease in FEV1 and normal or decreased forced vital capacity (FVC). The flow-volume curve shows a notch in the expiratory portion leading to a high decrease in flow with progressively lower volumes. Most patients do not have a positive bronchodilator response,28 although there may be moderate reversibility in up to 49% cases (≥12% and ≥200ml in FEV1).121,122 There may also be reversibility in 12.5% of patients with normal FEV1, suggesting that hyperreactivity may be an early characteristic of this disease.121
In the later stages of the disease, loss in the lung elastic recoil will result in an increased pulmonary compliance with hyperinflation, which translates into a decrease in FVC and an increase in total lung capacity (TLC) and in residual volume (RV). Hence, adding measurement of the pulmonary volumes to spirometry will allow for better and more complete patient assessment. Due to the effect of air entrapment, the pulmonary volumes measured by plethysmography are usually greater than those measured using helium-dilution based techniques28; therefore, they should be measured preferably using the first method.
Measuring carbon monoxide diffusing capacity (DLCO) will assess gas exchange through the alveolar-arterial membrane, which is usually reduced. Although these two aspects belong to the same process (emphysema), FEV1 and DLCO do not always correlate.28 Arterial blood gas analysis also enables us to assess gas exchange. There may not be any changes in the early stages, but as the disease progresses, hypoxemia will appear with exercise and then while resting, and will later on be associated with hypercapnia.
In the later stages of the disease the effect of emphysema on the muscular activity of the chest and diaphragm may be assessed by measuring inspiratory and expiratory muscle pressures.
Respiratory function in exercise may be assessed by means of exercise tests such as the six-minute walk test or the cardio-pulmonary exercise test. In later stages of the disease, cardio-pulmonary exercise test may show a decrease in the PaO2 and an increase in the alveolar-arterial difference. Patients suffering from AATD may have increased respiratory rates while resting and their minute ventilation may exceed 80% of predicted maximum voluntary ventilation during mild exercise, which is an indication that ventilation may limit higher levels of exercise.28 The 6-minute walk test assesses not only the exercise functional capacity, but it is also important to estimate disease prognosis by calculating the BODE index.
Imaging ExamsChest X-ray should be carried out during the initial assessment of all patients. However, high-resolution CT of the chest in order to assess the bronchopulmonary morphology or chest CT with thicker sections for densitometry are the best exams for detecting and quantifying the presence of emphysema.
Chest X-rayChest X-ray is the norm in the early stages of the disease. As the disease progresses the typical signs of hyperinflation, such as an increase in the anteroposterior and lateral diameter of the chest, higher transparency of the pulmonary fields, a decrease in the vascular markings, horizontalization of the ribs, an increase in the intercostal spaces, low and rectified hemidiaphragm, and vertical heart, appear. These changes are usually more prominent in the lower lung fields.28 Bullous changes do not occur very often in AATD and when they do occur (35%) their most likely location is in the inferior lobes.123
Chest CTChest CT is the best exam in vivo for characterising the severity and morphology of pulmonary emphysema,124 better than a chest X-ray or respiratory function tests.28 It has a positive correlation with pathological findings, and that is why it has been suggested as the best method for monitoring emphysema progression.101 The possibility of measuring pulmonary density enables quantification of the extent of emphysema which can be used to monitor disease progression.
In AATD, the emphysema is usually of the panlobular type28 and in 2/3 patients it particularly affects the basal regions, in contrast to the centrilobular emphysema prevailing in the upper lobes which is typical of emphysema caused by smoking. However, in up to 36% of the cases the emphysema may be diffuse or eventually apical.55
In CT, the emphysema areas are characterised by a particularly low attenuation, in contrast to the surrounding parenchyma in density values in Hounsfield units (HU). Chest CT provides for calculation of densitometry parameters, making it possible to assess the extent of the emphysema. There are several methods for calculating these densitometric parameters: the “density mask”, the “percentile method”, and the “average parenchyma density”. The density mask method uses a cut-off value (usually -910HU) and all pixels with density below this value will be classified as emphysema and compared in percentage to the entire lung parenchyma volume. The percentile method quantifies the emphysema through the cut-off point, which defines a percentile datum from the histogram. For example, the 10th percentile density (PD10) is derived from a histogram recording the densities in HU of all lung voxels and is defined as the threshold value for which 10% of all lung voxels have a lower density. As a true density measure this value will decrease as emphysema worsens. In the average density method, the average pulmonary parenchyma density is calculated, which will be lower as the emphysema progresses.124
Lung attenuation values (in HU) can be converted to lung tissue density values (g/l) by adding 1000 to the HU. For example, a 15th percentile density value of -950 HU equals a lung density of 50g/l, meaning that 15% of the pixels/voxels have a density value below 50g/l.125
In chest CT, other changes besides the emphysema may be seen, such as the thickening of the bronchial walls and bronchiectasis (25%) with a widening of the bronchial diameter at the segmental and sub-segmental level. When present, bronchiectasis are cylindrical or saccular and prevail in the lobes where emphysema is more extensive.101
Liver assessmentAll patients carrying alleles leading to AAT accumulation in the liver (Z, MMalton, MDuarte, SIiyama, etc.) must undergo an initial assessment of liver changes by means of abdominal ultrasound and serum tests.76
The serum liver assessment should include transaminases (AST and ALT) as well as alkaline phosphatase, GGT, bilirubin, albumin, coagulation tests, platelets, fat soluble enzymes, and alpha-fetoprotein.76,79 An abdominal ultrasound will evaluate liver changes – steatosis, portal hypertension, and cirrhosis – and the presence of hepatocellular carcinoma.76 FibroscanTM, a noninvasive technique using a probe with an ultrasound transducer and a mechanical vibrating device, exerts dynamic stress on the body surface to generate shear waves. The shear wave velocity can then be converted into liver stiffness, which is expressed in kilopascals. It may be used as an alternative to ultrasound to assess steatosis and liver fibrosis in patients with AATD.126
Liver biopsy should not be used for diagnosis, since the pathological changes vary and are not specific. The diagnostic method should be phenotyping or genotyping. However, liver biopsy may be indicated in certain cases, particularly when it becomes necessary to assess the level of liver damage, its progression, or to investigate the presence of other related diseases. There is usually lobe inflammation, variable hepato-cellular necrosis, fibrosis, cirrhosis, steatosis, and PAS-positive diastase-resistant globules in some, but not all, hepatocytes.76
- -
Initial evaluation of patients with AATD should include: spirometry, lung volumes, DLCO, arterial blood gas, chest X-ray, chest CT scan (with emphysema index analysis if available), 6-minute walk test
- -
In phenotypes with liver involvement, liver functions tests and abdominal ultrasound (or FibroscanTM) should be done.
[Research studies in the 1980s recognised that plasma α1-antitrypsin levels could be restored by intravenous infusions of purified human protein. Initial licensing of augmentation therapy in 1987 was based upon an assumption that its success in boosting circulating and lung levels of α1-AT would be of clinically meaningful benefit and a logical approach to long-term medical treatment. This assumption has subsequently been explored, and robust demonstration of such outcomes has proven problematic in trials (sensitive measures of lung destruction v. clinical efficacy)].127,128
Several scientific societies recommend, based on the available clinical evidence, the use of augmentation therapy, although with some differences in the indications for treatment as a result of the evaluation of clinical studies and date of publication. Based on a study published in 2003129 on the cost-effective assessment of treatment, the revised ERS guidelines,127 report a large increase in costs for relatively little increase in maintaining patient quality of life. Moreover, ERS guidelines also highlight that fact that the cost-benefit implications of the apparent reduction in exacerbation frequency and severity remain to be quantified.127
In Portugal, health authorities recognize the importance of patients to have access to augmentation therapy; however, the appreciation of medical evidence supporting the efficacy of this therapy by different prescribers and medical centres is still under discussion and may vary, causing inequalities in terms of clinical requirements for the approval of the therapy.
AUGMENTATION THERAPY WITH AAT[International expert treatment guidelines indicate that optimal management of Alpha -1 should include strict lifestyle standards, including smoking cessation, exercise, diet and symptomatic treatment with regular review with augmentation therapy intended to slow down progression and increase the chances of preventing irreversible tissue damage].130
The augmentation therapy available for AATD consists of the administration of an intravenous infusion of purified human AAT extracted from a pool of donor plasma. The main rationale for this therapy is to modify the course of the disease by preventing progression of the emphysema, reducing the number of exacerbations, and improving quality of life. In 1990, the observation that non-smoking individuals with SZ phenotype showed a low risk of developing pulmonary disease led to the general acceptance that AAT serum levels found in these individuals could represent a minimum protective threshold.131 Later, Campbell132 demonstrated that the mean area of damage exerted by neutrophils was unrelated to AAT concentration, until its level dropped below 10μM. Since then, the target for circulating AAT to give proper lung protection has been widely accepted to be 11μM (57mg/dl as determined by nephelometry).
The grounds for using AAT in the treatment of pulmonary disease are based on its biochemical role and clinical efficacy. Its intravenous administration resulted in an increment of AAT serum level above the protective threshold, increased concentration in the alveolar lining fluid, and neutralised activity of the serum and alveolar elastase from neutrophils throughout the interval between doses.133–136 A decrease in the degradation of elastase has also been shown through the quantification of serum, urinary and alveolar lining fluid levels of desmosine and isodesmosine markers.137,138
Stockley demonstrated a decrease in leukotriene B4 (LTB4), myeloperoxidase (MPO) and interleukin 8 (IL8), albeit only significantly reduced in patients undergoing AAT augmentation therapy.136
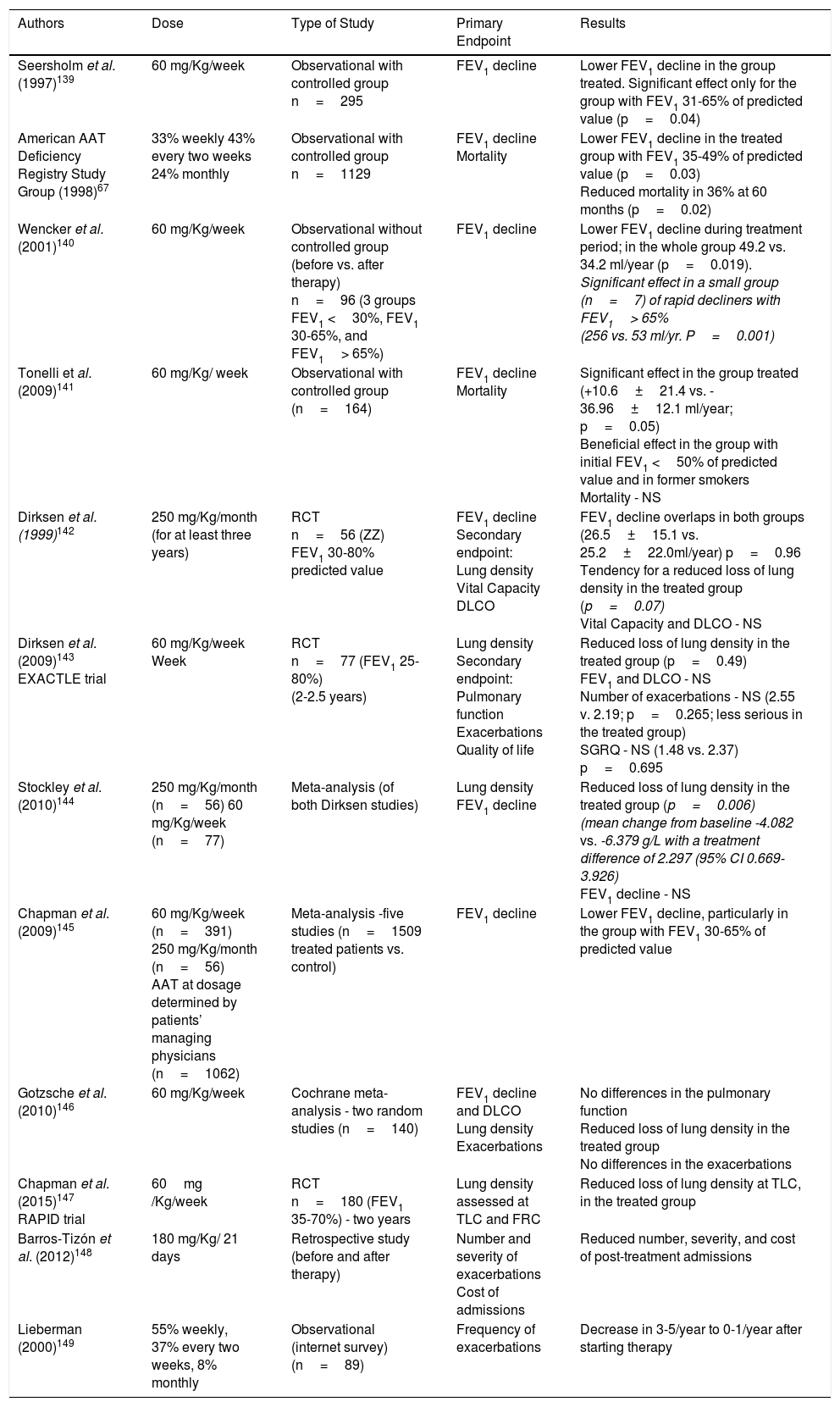
Clinical efficacy should address the following questions: does AAT augmentation therapy have a positive effect in lowering the decline of pulmonary function and in delaying the progression of emphysema? Does it decrease the number and severity of exacerbations? Does it improve functional capacity, patient symptoms and survival? Is it safe and cost-effective?8 Hence, medical grounds are based on the results of various studies with different designs (observational or randomized and placebo-controlled) comparing the decline in pulmonary function, variation of lung density assessed by chest CT, mortality rate, frequency and severity of exacerbations, impact on the quality of life and functional capacity between groups of patients submitted to therapy and patients who have not been treated. Among these, the randomized studies controlled with placebo stand out, since they have demonstrated a smaller loss in lung density quantified by CT in subjects that have been submitted to AAT therapy. Table 5 summarizes the main results for studies supporting the use of the AAT therapy.
Studies carried out over the years to evaluate efficacy of AAT augmentation therapy.
| Authors | Dose | Type of Study | Primary Endpoint | Results |
|---|---|---|---|---|
| Seersholm et al. (1997)139 | 60 mg/Kg/week | Observational with controlled group n=295 | FEV1 decline | Lower FEV1 decline in the group treated. Significant effect only for the group with FEV1 31-65% of predicted value (p=0.04) |
| American AAT Deficiency Registry Study Group (1998)67 | 33% weekly 43% every two weeks 24% monthly | Observational with controlled group n=1129 | FEV1 decline Mortality | Lower FEV1 decline in the treated group with FEV1 35-49% of predicted value (p=0.03) Reduced mortality in 36% at 60 months (p=0.02) |
| Wencker et al. (2001)140 | 60 mg/Kg/week | Observational without controlled group (before vs. after therapy) n=96 (3 groups FEV1 <30%, FEV1 30-65%, and FEV1> 65%) | FEV1 decline | Lower FEV1 decline during treatment period; in the whole group 49.2 vs. 34.2 ml/year (p=0.019). Significant effect in a small group (n=7) of rapid decliners with FEV1> 65% (256 vs. 53 ml/yr. P=0.001) |
| Tonelli et al. (2009)141 | 60 mg/Kg/ week | Observational with controlled group (n=164) | FEV1 decline Mortality | Significant effect in the group treated (+10.6±21.4 vs. - 36.96±12.1 ml/year; p=0.05) Beneficial effect in the group with initial FEV1 <50% of predicted value and in former smokers Mortality - NS |
| Dirksen et al. (1999)142 | 250 mg/Kg/month (for at least three years) | RCT n=56 (ZZ) FEV1 30-80% predicted value | FEV1 decline Secondary endpoint: Lung density Vital Capacity DLCO | FEV1 decline overlaps in both groups (26.5±15.1 vs. 25.2±22.0ml/year) p=0.96 Tendency for a reduced loss of lung density in the treated group (p=0.07) Vital Capacity and DLCO - NS |
| Dirksen et al. (2009)143 EXACTLE trial | 60 mg/Kg/week Week | RCT n=77 (FEV1 25-80%) (2-2.5 years) | Lung density Secondary endpoint: Pulmonary function Exacerbations Quality of life | Reduced loss of lung density in the treated group (p=0.49) FEV1 and DLCO - NS Number of exacerbations - NS (2.55 v. 2.19; p=0.265; less serious in the treated group) SGRQ - NS (1.48 vs. 2.37) p=0.695 |
| Stockley et al. (2010)144 | 250 mg/Kg/month (n=56) 60 mg/Kg/week (n=77) | Meta-analysis (of both Dirksen studies) | Lung density FEV1 decline | Reduced loss of lung density in the treated group (p=0.006) (mean change from baseline -4.082 vs. -6.379 g/L with a treatment difference of 2.297 (95% CI 0.669-3.926) FEV1 decline - NS |
| Chapman et al. (2009)145 | 60 mg/Kg/week (n=391) 250 mg/Kg/month (n=56) AAT at dosage determined by patients’ managing physicians (n=1062) | Meta-analysis -five studies (n=1509 treated patients vs. control) | FEV1 decline | Lower FEV1 decline, particularly in the group with FEV1 30-65% of predicted value |
| Gotzsche et al. (2010)146 | 60 mg/Kg/week | Cochrane meta-analysis - two random studies (n=140) | FEV1 decline and DLCO Lung density Exacerbations | No differences in the pulmonary function Reduced loss of lung density in the treated group No differences in the exacerbations |
| Chapman et al.(2015)147 RAPID trial | 60mg /Kg/week | RCT n=180 (FEV1 35-70%) - two years | Lung density assessed at TLC and FRC | Reduced loss of lung density at TLC, in the treated group |
| Barros-Tizón et al. (2012)148 | 180 mg/Kg/ 21 days | Retrospective study (before and after therapy) | Number and severity of exacerbations Cost of admissions | Reduced number, severity, and cost of post-treatment admissions |
| Lieberman (2000)149 | 55% weekly, 37% every two weeks, 8% monthly | Observational (internet survey) (n=89) | Frequency of exacerbations | Decrease in 3-5/year to 0-1/year after starting therapy |
Abbreviations: RCT, randomized controlled trial; NS, non-significant
Age – the age range for starting treatment is unknown; however, most patients are expected to be in the fourth decade of life when diagnosed, since the emphysema usually develops approximately ten years earlier than in smoking-related COPD. The minimal age for initiating therapy is 18 and any exception must be justified. The maximum age limit may be established depending on each case. Table 6
Criteria for augmentation therapy.
| Non-smokers or ex-smokers (≥ 6 months) |
| Age ≥ 18 |
| COPD diagnosis (FEV1/FVC <70 postbronchodilator) attributed to emphysema caused by AAT deficiency |
| Serum level of AAT ≤ 57 mg/dl |
| No selective immunoglobulin A deficiency |
| FEV1 30-70% of predicted value |
| FEV1> 70% in the case of rapid decliner (decrease> 120 ml/year) |
| Individual decision in other cases |
| Augmentation therapy should not be discontinued in case of pulmonary function deterioration, even if it reaches the lower established limit for its initiation |
Panniculitis: Consider treatment if recurrent and low ATT serum level
Genotype - pulmonary disease in AATD is associated with genotypes such as ZZ, ZQ0, Q0Q0, and other combinations with rare deficient or null variants. The clinical efficacy of the AAT therapy is better documented in the treatment of ZZ patients.
Importantly, knowing that approximately 10% of patients with SZ genotype have a serum level below the protective threshold, it seems acceptable to include these subjects, as long as all other criteria for therapy are verified. However, for SZ individuals there is no scientific evidence supporting clinical efficacy of the therapy.
AAT Level – Decisional cut-off - Patients with serum levels ≤ 57mg/dl (nephelometry) are candidates for AAT replacement therapy. Table 7
Procedures before initiating therapy.
| Complete liver function tests and liver ultrasound |
| Serum immunoglobulin tests |
| Hepatitis (B and C) and acquired human immunodeficiency virus serologies |
| Hepatitis B vaccination |
| Pulmonary Function Testing - Spirometry, Pulmonary Volumes, and Carbon Monoxide Diffusing Capacity |
| Arterial blood gas analysis and 6-minute walk test |
| High resolution chest CT and assessment of lung density (if available) |
Pulmonary Functional assessment and CT Densitometry
Observational studies indicate a smaller decline in pulmonary function of patients undergoing therapy with airways obstruction within FEV1 31-65% or FEV1 35-49% of predicted values. Patients with more severe obstructions do not appear to have benefitted from therapy.67,139 A study carried out in 2009 and comprising 164 patients, concluded that a beneficial effect was observed in the group of ex-smokers with FEV1 <50% predicted value.141 Furthermore, in a meta-analysis that included five clinical trials and 1509 patients undergoing therapy vs. controlled patients, the treated group registered a lower functional decline with this result showing greater significance in the group of patients with FEV1 between 30-65% of predicted value.145
The analysis of these studies suggests that the decline in FEV1 is not linear and drops faster in the moderate obstruction interval.150 Nevertheless, Wencker140 carried out a multi-centre and retrospective study to assess the progress of emphysema before and during the AAT therapy (assessment of the 47 months prior and the 50 months during therapy – in the 60mg/Kg/week dose), which included 96 patients split into three functional groups – FEV1 <30%, between 30-65% and> 65%, and they verified the existence of patients with severe deficiency, preserved pulmonary function (FEV1> 65%) and fast declining pulmonary function (> 120ml/year). Moreover, in this latter group, the loss of pulmonary function prior and during treatment was 255±70.4 and 52.7±61.3ml, respectively and according to these authors, an early implementation of AAT therapy significantly reduced patient decline of respiratory function.
More recently, randomized studies controlled with placebo, in which the emphysema progression is quantified by CT scans, have proposed this technology as the best indicator of the efficiency of this therapy. The authors find that CT densitometry is more sensitive than the variation of physiological indicators or quality of life143 as there may be loss of lung density with stable FEV1 values.150 In an initial study, which had as its primary outcome the assessment of the FEV1 decline, 56 patients with FEV1 between 30-80% were randomised for a dose of 250mg/Kg/4 weeks, for three years. No significant difference in the decline of the pulmonary function was detected between patients although there was a tendency for a reduced loss of lung density in the active treatment group.142
The EXACTLE study143 lasted for 24-30 months and included 77 patients with postbronchodilator FEV1 between 25-80% for the dose of 60mg/Kg/week vs. placebo. The endpoint goal of this study was to analyse the loss of lung density by CT measures. A marginal statistical significance was observed in only one of the four analyses performed supporting the AAT treatment as beneficial. Another objective of EXACTLE was to evaluate the frequency and severity of exacerbations among groups of patients. Only the number of hospital admissions for exacerbation was lower in the AAT treated group due to a reduction in exacerbation severity. In addition, there was also found a correlation between the decline of pulmonary function and the decline of lung density, despite no difference being observed in the functional decline rate between groups. An integrated assessment of the findings in these two studies showed a significant reduction in the decline of lung density in treated patients, where -2.297g/L (p=0.006) difference was found between groups.144
The RAPID study,147 which used the analysis of lung density as a primary endpoint and included 180 patients with FEV1 between 35-70%, showed that augmentation therapy contributes to the preservation of lung density when assessed in terms of TLC. This study lasted two years and patients were split into two groups (active treatment group – AAT at the dose of 60mg/Kg/week vs. placebo). Lung density loss of 1.45g/L/year was observed in the treated group vs. 2.19g/L/year in the placebo group with significant difference (p=0.03). However, no differences were found between the groups in terms of FEV1, carbon monoxide diffusing capacity, results on the shuttle test, and health-related quality of life improvement. This study also specified that treatment benefits were dose-related and more expressive in patients with higher serum trough AAT concentration. The dose of 60mg/Kg/week was not established as an optimal dose. Higher trough concentrations were achieved during treatment in heavier patients with greater AAT concentrations in pre-treatment.147
An open label period followed (RAPID Extension-2 years), showing that when patients of the placebo group began treatment with AAT, their lung density decline rate would overlap that of the actively treated group although the lost density would not be recovered, which suggested a need for early treatment. However, the authors stated that they did not know whether the preservation of lung density or structure was uniform to all deficient patients or in every stage of the disease.147
The results of these three studies described above, which included mostly ZZ patients, were presented without discrimination of any functional subgroup where the decrease in lung density decline rate could have been slower; therefore leaving the impression that those patients with FEV1 in the established intervals for the studies (FEV1 between 25/30/35 – 70/80% of predicted value) would benefit from the AAT therapy. The ATS/ERS document28 provides the possibility of treating patients with an almost normal pulmonary function as long as they are identified as rapid decliners and the Canadian151 and Spanish Pneumology Societies110 recommend, respectively, AAT treatment for patients with FEV1 between 25-80% and FEV1 <80% of predicted value.
This controversy regarding treatment of patients with FEV1>65% was approached in a review article published by Teschler in March 2015 with the recommendation based on the author's experience to functionally assess these patients every six months. Rapid decliners may initiate therapy although this is an individual decision.152
In brief, the collected evidence points towards AAT therapy being efficient in preserving lung density although there are no significant results regarding other indicators, such as health-related quality of life and/or exacerbations. Regarding functional decline, results are contradictory, with some observational studies and a meta-analysis suggesting a smaller decline in the group of patients with moderate bronchial obstruction. Randomized studies did not find a significant difference in the FEV1 decline rate between the treatment group and the placebo group. In relation to other indicators studied such as mortality, the American Registry67 has revealed a reduction in the group of treated patients with FEV1 <50% while the study led by Tonelli141 did not show any differences between the two groups.
Taking into account all these data, the Portuguese study group of AATD reached an agreement to recommend AAT augmentation therapy be provided to all patients with FEV1 between 30-70% of predicted value or, if higher, in the case of a rapid decline. Concerning treatment of liver disease, the augmentation therapy is only indicated in subjects with concomitant pulmonary involvement. In other cases only preventive measures are available (vaccination anti-A and B hepatitis, elimination of alcohol and other hepatotoxic agents).153 Still, AAT augmentation therapy may be indicated for the treatment of necrotising panniculitis due to its efficacy in some clinical cases and in the prevention or control of skin lesions related to this disease.154,155
Selective immunoglobulin A (IgA) deficiency is a contraindication to treatment as augmentation therapy may contain small amounts of IgA. Hypersensitive or anaphylactic reactions with anti-IgA antibodies may occur in these patients, therefore, testing for this deficiency is recommended before initiating therapy.156
OTHER THERAPIESLung volume reduction surgery in patients with severe pulmonary emphysema has shown modest results in terms of survival and functional capacity in patients with emphysema with upper lobe predominance; experience with patients with AATD is limited but there seems to be a tendency to greater mortality within this subgroup.157 Lung volume reduction by placing endobronchial valves is an experimental intervention in patients with severe emphysema and AATD was an exclusion criterion in the biggest randomized study published.158,159
Pulmonary transplantation is reserved for patients with no other choice of therapy and the Portuguese group is following the current international recommendations. AATD is an indication for transplantation in about 5.8% of cases. Patients with no contraindications and a BODE index>5 may be referred to a pre-transplant consultation. Indications for combined liver and lung transplantation are rare but may be considered when there is documented cirrhosis in biopsy and a portal gradient> 10mmHg160,161. There is no evidence for recommending augmentation therapy after pulmonary transplantation; although some authors recommend considering it in case of infection or acute rejection. In selected cases with accelerated functional deterioration one might consider restarting augmentation therapy and maintaining it based on results.152,156
AVAILABLE FORMULATIONSThe following formulations are currently available in Portugal: Prolastin® and Respreeza®.162
Dosage plans: The current plan approved by the FDA (Food and Drugs Administration) is weekly administration (60mg/Kg), since it was one of the first to be proposed, the one analysed the most, and recommended by ATS/ERS.28,79 However, for the sake of cost reduction and convenience for the patient, other dosages have been studied but with no formal recommendation for any change in dose or administration interval. The doses of 50mg/kg/7 days and 120mg/kg/14 days have appeared to maintain Cmin higher than those considered as protective in 90% of patients.163–166 The dose of 180mg/Kg/21 days is able to maintain a protective Cmin for 85% of the time between doses.163,166 Hubbard and Dirksen studied monthly dosages, where 250mg/kg of AAT was administered with serum levels and anti-elastase activity showing values considered as protective for 28 days following infusion.135,142
Efficacy and safety: Experience with augmentation therapy suggests that it generally is safe, well tolerated and with very few adverse effects. In the study carried out by Wencker, who followed 443 patients over the course of six years, it was noted that 86% of patients did not have any adverse reactions and the following were the most common effects reported: nausea and vomiting (4.7%), hives (4.1%), fever (3.8%), dyspnoea (3.8%) and anaphylactic reaction (0.9%).167 In the study carried out by Stoller, with 747 patients over the course of seven years, it was noted that 83% of patients did not have any adverse reactions. The most common adverse effects were headaches (47%), dizziness (17%), nausea (9%), and dyspnoea (9%).168 Taking into account the fact that this product is derived from human plasma, there appears to be no case where prior disease, human immunodeficiency virus (HIV), viral hepatitis, or the development of viral antibodies were transmitted.133,169 It should be noted that the studies have shown a lower rate of adverse effects in patients who were submitted to monthly therapy than those who were submitted to weekly therapy.169
AAT administration by aerosol may be a promising option among the several therapy alternatives being studied. It is immediately directed to the target (lung) bypassing the short plasma half life limitations. Assuming that smaller quantities of the drug may be required, therapy by aerosol may be cheaper in addition to allowing self-administration and enabling production of large quantities with no risk of viral contamination.150
PATIENT FOLLOW-UPAATD patient follow-up may be considered on two levels: before and after initiating replacement therapy. Individuals who were diagnosed in their childhood may never have any complaints if they never smoke. A respiratory functional assessment is suggested at the end of adolescence and afterwards at 2-3 year intervals. Data on the decline of the pulmonary function will always be useful for comparison purposes in case of eventually initiating therapy.170 In the case of patients with no criteria for initiating therapy at the time of diagnosis – AAT concentration below the recommended threshold with no functional criteria –, follow-up should consist of spirometry every semester, annual assessment of liver function and chest CT with emphysema quantification (if available), without prejudice for individual decision at all times.152
The follow-up of patients undergoing augmentation therapy should include79,156: spirometry with bronchodilator test every semester; plethysmography and the study of carbon monoxide diffusion capacity once a year. Although ERS/ATS and SEPAR recommend an annual assessment of liver function, an efficient screening of liver disease will imply blood tests – complete blood count, liver enzymes – and abdominal ultrasound. In the case of individuals with an established liver disease, ultrasound, liver function, and dosage of alpha-fetoprotein should be repeated at regular intervals every six to 12 months.171 High resolution chest tomography is not recommended in follow-up although it may be repeated in order to clarify any complications. Lung density measurements will be of particular interest within the scope of medical research. Hence, assessment before replacement will mostly be of pulmonary function although later CT with densitometry appears to be the most sensitive indicator for assessing benefits to the patient.
- 1.
In the therapeutic approach of patients with emphysema due to AATD, international COPD recommendations should be followed for smoking cessation, symptomatic treatment, referral to respiratory rehabilitation programmes, exercise, diet, and vaccination.
- 2.
Recent studies have shown clinical efficacy of augmentation therapy in preserving pulmonary density, quantified by CT.
- 3.
Criteria for augmentation therapy are: age ≥ 18; non-smokers or ex-smokers (≥ 6 months); AAT ≤ 57 mg/dl (nephelometry); deficient phenotypes (ZZ, ZQ0, Q0Q0, SZ, or other allele combinations with rare variants); COPD diagnosis (FEV1/FVC <70) attributed to emphysema caused by AAT deficiency and FEV1 postbronchodilator between 30-70% of that predicted; no immunoglobulin A deficiency; augmentation therapy is not indicated in liver disease (if there is no pulmonary involvement), and it may be considered in panniculitis.
- 4.
Dose: 60 mg/kg/ week.
- 5.
AAT serum level testing is not recommended for therapeutic monitoring.
- 6.
Follow-up: 1) respiratory monitoring: spirometry with bronchodilator test per semester/year; annual plethysmography and carbon monoxide diffusion capacity; chest CT – at the initial diagnosis and repeatedly as per medical indication. 2) Liver monitoring: annual laboratory assessment; abdominal ultrasound – every 6 to 12 months.
- 7.
Lung volume reduction surgery is not recommended in this group of patients
- 8.
There are precise indications for referral to pulmonary transplantation.
This article is part of a supplement entitled “Portuguese consensus document for the management of alpha-1-antitrypsin deficiency” which is sponsored by Sociedade Portuguesa de Pneumologia.
Conflicts of interestF. Costa reports personal fees and non-financial support from Grifols and personal fees from CSL Behring, outside the submitted work; J Gomes reports personal fees from CSL Behring and non-financial support from Grifols, outside the submitted work; D. Maia reports non-financial support from CSL Behring and from Grifols Portugal, outside the submitted work; M.A. Mineiro, L. Telo, C. Santos, C. Antunes, M. Canotilho, E. Magalhães, I. Vicente, C. Valente, B.G. Gonçalves, B. Conde, C. Guimarães, C. Sousa, J. Amado, M.E. Brandão, M. Sucena, M.J. Oliveira, S. Seixas, V. Teixeira, A.P. Lopes, R. Melo have nothing to disclose.


