




Pompe disease is a rare autosomal recessive neuromuscular disorder caused by acid α-glucosidase enzyme (GAA) deficiency and divided into two distinct variants, infantile- and late-onset. The late-onset variant is characterized by a spectrum of phenotypic variation that may range from asymptomatic, to reduced muscle strength and/or diaphragmatic paralysis. Since muscle strength loss is characteristic of several different conditions, which may also cause diaphragmatic paralysis, a protocol was created to search for the diagnosis of Pompe disease and exclude other possible causes.
MethodsWe collected a sample size of 18 patients (10 females, 8 males) with a median age of 60 years and diagnosis of diaphragmatic paralysis of unknown etiology, followed in the Pulmonology outpatient consultation of 9 centers in Portugal, over a 24-month study period. We evaluated data from patient's clinical and demographic characteristics as well as complementary diagnostic tests including blood tests, imaging, neurophysiologic and respiratory function evaluation. All patients were evaluated for GAA activity with DBS (dried blood test) or serum quantification and positive results confirmed by serum quantification and sequencing.
ResultsThree patients were diagnosed with Pompe's disease and recommended for enzyme replacement therapy. The prevalence of Pompe, a rare disease, in our diaphragmatic paralysis patient sample was 16.8%.
ConclusionWe conclude that DBS test for GAA activity should be recommended for all patients with diaphragmatic paralysis which, despite looking at all the most common causes, remains of unknown etiology; this would improve both the timing and accuracy of diagnosis for Pompe disease in this patient population. Accurate diagnosis will lead to improved care for this rare, progressively debilitating but treatable neuromuscular disease.
Pompe disease is a rare autosomal recessive neuromuscular disorder caused by acid α-glucosidase enzyme (GAA) deficiency, resulting in the accumulation of glycogen in the lysosomes of numerous, but primarily muscular, tissues.1 It is often termed glycogen storage disease type II (GSDII) or acid maltase deficiency.2,3
Depending on the age of onset, Pompe is divided into infantile and late-onset4 disease. Infantile onset Pompe disease represents the most severe form and almost invariably leads to death, due to cardio-respiratory failure, within one year. The late-onset variant presents at any time after the age of one year, and is characterized by a spectrum of phenotypic variation. It may range from asymptomatic patients with increased creatine kinase (CK) to muscle cramps and pain syndrome or rigid-spine syndrome.5–7 The lower limbs and paraspinal muscles are frequently affected first, followed by the respiratory muscles, particularly the diaphragm, intercostal and accessory muscles. Respiratory failure is the main cause of increased morbidity and mortality8,9 and the main cause of respiratory failure is diaphragmatic weakness.10
The diaphragm is the major muscle of ventilation constituted by a dome-shaped structure of tendons and muscle innervated by the phrenic nerves which provide sensory, sympathetic and motor function. Diaphragm contraction expands the chest increasing pleural negative pressure and promoting air flow into the lungs. Whenever there is diaphragmatic weakness the diaphragm fails to contract appropriately, causing reduced inspiratory volume and possible dyspnea. An extreme form of diaphragmatic weakness is unilateral or bilateral diaphragmatic paralysis.11 Several different conditions may present with respiratory failure and distinguishing among them is important to determine the correct therapeutic options.12 Pompe disease is in the differential diagnosis of a wide variety of myopathies, and therefore accurate diagnostic tools are needed for an effective screening of this condition. Particularly when the progressive involvement and weakness of respiratory muscles and diaphragm, characteristic of the disease, are closely related to respiratory dysfunction which affects sleep, daily life activities, and overall quality of life.
The prevalence of this condition in the general population is unknown, and varies according to clinical presentation and ethnicity. The late-onset form has an estimated incidence of 1/57000.13 In Portugal, there is no accurate data on the prevalence of Pompe disease, but it is estimated that there are 27 cases of patients under enzyme replacement therapy (ERT).14–16
Therefore, the primary objective of this study was to estimate the prevalence of this disorder in patients with a diagnosis of diaphragmatic paralysis of unknown etiology followed in the outpatient setting of a Pulmonology clinic, and to characterize the clinical and socio-demographic profile of these patients. A secondary objective was to create an algorithm for the accurate diagnosis of late-onset Pompe disease in patients with diaphragmatic paralysis of unknown etiology, which will lead to better management and treatment of patients with this neuromuscular disease.
Materials and methodsThis was a national, multicenter, epidemiological study of patients with a diagnosis of diaphragmatic paralysis of unknown etiology. The study population was identified from the Pulmonology outpatient consultations of 9 centers in Portugal over a 24-month study period. Patients were consecutively enrolled if they were ≥18 years of age, gave informed consent to participation, and fulfilled one of the following inclusion criteria: diagnosis of (unilateral or bilateral) diaphragmatic paralysis of unknown cause; diagnosis of restrictive lung disease (both FVC [forced vital capacity] and TLC [total lung capacity]<80% of predicted, or ≥12% decrease in VC [vital capacity] in the supine position), decreased maximal inspiratory pressure (IPmax) or sniff nasal inspiratory pressure (SNIP) (at least −10cm of H2O than predicted) of unknown origin; diagnosis of progressive chronic myopathy with respiratory involvement, particularly of the diaphragm, with either inconclusive muscle biopsy or a diagnosis based on the generic definition of inclusion body myositis.
Patients were excluded from the study if they were unable or unwilling to provide informed consent, were tracheostomized or pregnant, required invasive mechanical ventilation, or suffered from one of the following conditions: neuromuscular junction diseases (Eaton–Lambert syndrome, myasthenia gravis), myopathies (polymyositis and other mixed connective tissue diseases, dystrophy mitochondrial myopathies, amyloidosis), spinal cord myelopathy (cervical spine injury, sarcoidosis, syringomyelia, polyomyelitis, amyotrophic lateral sclerosis), peripheral neuropathy [cervical spine injury, mediastinal tumor, Guillain–Barré syndrome, nutritional neuropathies (vitamin B12 deficiency) and lead neuropathy] or active neoplastic disease.
Information retrieved from patients included socio-demographic and clinical history, as well as physical exams and diagnostic parameters (including respiratory function tests, polysomnography, ultrasound scan, chest radiograph and blood biochemistry), all collected as part of the routine clinical practice for diaphragmatic paralysis diagnosis. Acid α-glucosidase activity was determined in dried-blood spots (DBS) for the diagnosis of Pompe disease,17 and a further confirmation through GAA gene sequencing was done if the blood-based GAA enzyme assay showed reduced activity.18
Respiratory function testsRespiratory function tests were conducted using body plethysmography with vital capacity determination in seated versus supine position. The single-breath method was used to measure the alveolo-capillary diffusion of CO (DLCO-SB) and thoracic pressures were ascertained either by maximal inspiratory pressure (MIP) or by sniff nasal inspiratory pressure (SNIP) followed by maximal expiratory pressure (MEP).
Statistical analysisStatistical analysis was performed using SPSS 15.0 software. Continuous variables were described as medians and interquartile ranges, and categorical variables were described as numbers and (proportions with) percentages. A multivariate analysis was performed to evaluate the association between diaphragmatic paralysis and muscle strength loss characteristic of Pompe disease.
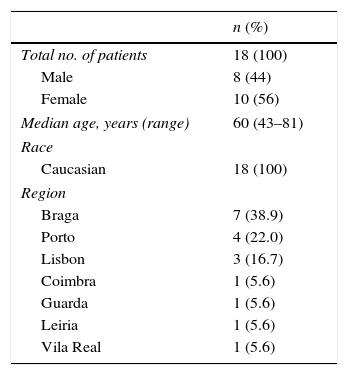
ResultsA total of 18 patients with diaphragmatic paralysis were included in the DIPPER study by the collaborative Portuguese centers. Patients’ socio-demographic baseline characteristics are reported in Table 1. Eight (44%) patients were male and 10 (56%) were female, the median age was 60.8±11.1 (range 43–81) years, all patients were Caucasian and mainly from the North of Portugal.
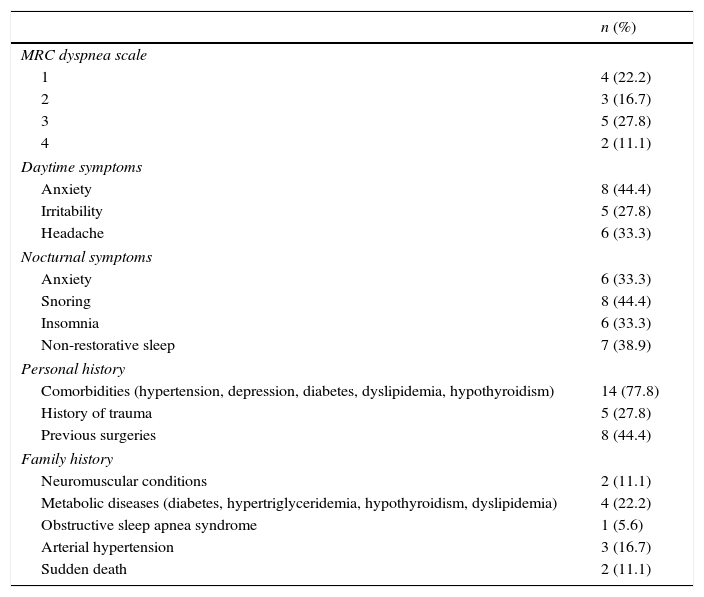
Patient clinical history is summarized in Table 2. Briefly, most patients (27.8%) were grade 3 (walks slower than most people on the level, stops after a mile or so, or stops after 15min walking at own pace) in the MRC dyspnea scale, and 57.1% experienced anxiety as the main daytime symptom. Excessive daytime sleepiness (EDS) was measured through the Epworth sleepiness scale (ESS) with a median value of 7 and an interquartile range of 4–13, the maximum value for the Epworth scale attained by any patient was 19. Several nocturnal symptoms were reported, snoring (61.5%) and non-restorative sleep (53.8%) being the most frequent. Arterial hypertension was the most frequent health precedent, and metabolic diseases the most frequent family precedent (n=4, 22.2%).
Patients’ clinical characteristics.
| n (%) | |
|---|---|
| MRC dyspnea scale | |
| 1 | 4 (22.2) |
| 2 | 3 (16.7) |
| 3 | 5 (27.8) |
| 4 | 2 (11.1) |
| Daytime symptoms | |
| Anxiety | 8 (44.4) |
| Irritability | 5 (27.8) |
| Headache | 6 (33.3) |
| Nocturnal symptoms | |
| Anxiety | 6 (33.3) |
| Snoring | 8 (44.4) |
| Insomnia | 6 (33.3) |
| Non-restorative sleep | 7 (38.9) |
| Personal history | |
| Comorbidities (hypertension, depression, diabetes, dyslipidemia, hypothyroidism) | 14 (77.8) |
| History of trauma | 5 (27.8) |
| Previous surgeries | 8 (44.4) |
| Family history | |
| Neuromuscular conditions | 2 (11.1) |
| Metabolic diseases (diabetes, hypertriglyceridemia, hypothyroidism, dyslipidemia) | 4 (22.2) |
| Obstructive sleep apnea syndrome | 1 (5.6) |
| Arterial hypertension | 3 (16.7) |
| Sudden death | 2 (11.1) |
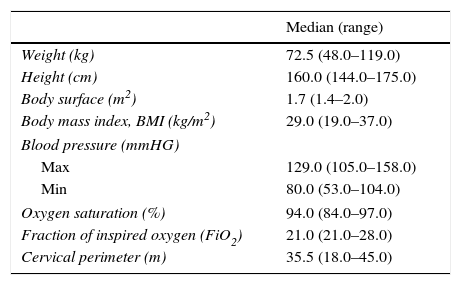
Overall physical status was assessed in 14 patients by empirical observation with 9 (64.3%) patients reported as having good physical function, and 5 (35.7%) reported to have modest physical function. Anthropometrics of these patients are depicted in Table 3.
Anthropometrics of the 14 patients who performed the physical examination.
| Median (range) | |
|---|---|
| Weight (kg) | 72.5 (48.0–119.0) |
| Height (cm) | 160.0 (144.0–175.0) |
| Body surface (m2) | 1.7 (1.4–2.0) |
| Body mass index, BMI (kg/m2) | 29.0 (19.0–37.0) |
| Blood pressure (mmHG) | |
| Max | 129.0 (105.0–158.0) |
| Min | 80.0 (53.0–104.0) |
| Oxygen saturation (%) | 94.0 (84.0–97.0) |
| Fraction of inspired oxygen (FiO2) | 21.0 (21.0–28.0) |
| Cervical perimeter (m) | 35.5 (18.0–45.0) |
The Mallampati score, used to predict the ease of intubation, was assessed in 13 patients and showed that most of them (n=5, 38.5%) were class 2 (soft palate, fauces, portion of uvula), 4 (30.8%) patients were class 3 (soft palate, base of uvula), 3 (23.1%) patients were class 1 (soft palate, fauces, uvula, pillars), and only 1 (7.7%) patient was class 4 (hard palate only).
Neurologic evaluation revealed that most patients had difficulty getting up (n=8, 61.5%) and sitting (n=7, 53.8%). Concerning upper limbs, cases of muscular atrophy were observed in 3 (23.1%) patients, mainly in arms, forearms and hands, 8 (72.7%) patients had sustained muscular strength, and 10 (76.9%) patients had normal reflexes. Concerning lower limbs, muscular atrophy was observed in 3 (23.1%) patients, 9 (69.2%) patients had sustained muscular strength, and 9 (69.2%) patients had normal reflexes. An abnormal gait was identified in 7 (58.3%) patients, mainly due to ataxia and weakness in both feet (n=2, 28.6%, each), but also due to Trendelenburg walk in one patient, paraparesis in one patient, and as a consequence of bimalleolar osteoporotic fracture in one patient. Nine (69.2%) patients were unable to run, with main causes being fatigue, dyspnea, and sequels from bimalleolar osteoporotic fracture.
Complementary diagnostic testsFifteen of the 18 patients included in this study performed complementary diagnostic tests, including complete blood count (CBC) and blood biochemistry (Table 4), blood serology and thyroid function assessment. For 88.9% of these patients, thyroid function was within the normal range. One patient had positive serology for HBV, and another for HCV. Immunoglobulins were within the normal range for 13 patients and were abnormal for two patients, and abnormal levels of autoantibodies were also observed in two patients.
Complementary diagnostic test results for the 15 patients who performed them.
| Median (range) | |
|---|---|
| CBC | |
| Hemoglobin (g/100ml) | 13.6 (10.1–16.8) |
| Leukocyte count | 6725 (1160–11400) |
| Neutrophil count | 3820 (824–4964) |
| Lymphocyte count | 1425 (209–1750) |
| Platelet count | 237 (160–2390) |
| Blood biochemistry | |
| Sodium (mmol/L) | 141 (135–147) |
| Creatinine (mg/dl) | 0.7 (0.5–5.5) |
| Potassium (mmol/L) | 4.4 (3.4–5.0) |
| Glucose (mg/DL) | 102.5 (81–156) |
| Protein (g/L) | 70 (58–75.4) |
| Albumin (g/L) | 42 (35–62.2) |
| Creatine kinase (CK) (IU/L) | 152 (24–1290) |
| Calcium (mmol/L) | 8.8 (2.3–9.9) |
| Phosphorus (mmol/L) | 3.2 (0.9–34) |
| Alkaline phosphatase | 63.5 (34–117) |
| Gamma-glutamyl transferase (GGT) (IU/L) | 28 (8–209) |
| Alanine aminotransferase (ALT) (IU/L) | 32 (6–105) |
| Aspartate aminotransferase (AST) (IU/L) | 25.5 (15–94) |
| Total bilirubin (μmol/L) | 9.2 (3.4–23.7) |
| Direct bilirubin (μmol/L) | 0.2 (0.18–0.21) |
| Total cholesterol (mg/dL) | 228.5 (119–319) |
| Thyroglobulin (ng/dL) | 105.5 (2.3–291) |
| Sedimentation velocity (S) | 10 (5–80) |
| Angiotensin-converting enzyme (ACE) (unit/L) | 15 (3–67) |
IU/L=international units/liters; S=svedberg unit.
Creatine kinase (CK) levels were elevated in these patients, with a mean CK of 249.8 (SD=332.9)U/L, and a median of 152 (range: 24–1290) U/L.
The chest X-ray revealed abnormal results in 13 patients, due to (right, left or both) hemidiaphragm elevation. In all patients diaphragmatic paralysis were confirmed by thoracic CT scan (Fig. 1) or thoracic ultrasound (Fig. 2) and changes in diaphragmatic movement ascertained.
Respiratory function tests
 and normal left diaphragmatic movement amplitude (B).")
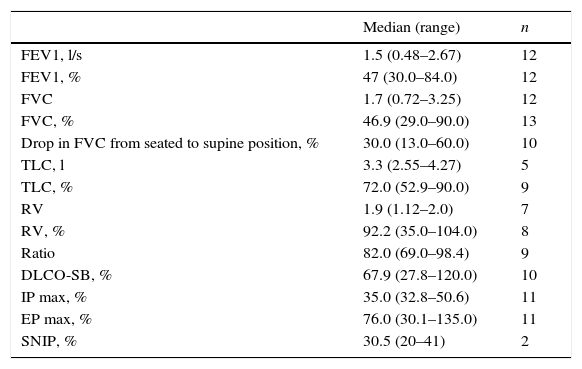
Respiratory function tests were mandatory to assess whether there was a deterioration of patients’ respiratory condition. They may be indicative, for instance, of diaphragmatic weakness by showing a difference between forced vital capacity (FVC) in sitting and supine position – i.e. postural drop – or by a decreased mean inspiratory pressure (MIP). Spirometry was used to measure how much and how quickly patients could move air out of their lungs, and lung function values obtained are shown in Table 5, as well as the number of patients in which this evaluation was collected.
Respiratory function tests (PFTs) results.
| Median (range) | n | |
|---|---|---|
| FEV1, l/s | 1.5 (0.48–2.67) | 12 |
| FEV1, % | 47 (30.0–84.0) | 12 |
| FVC | 1.7 (0.72–3.25) | 12 |
| FVC, % | 46.9 (29.0–90.0) | 13 |
| Drop in FVC from seated to supine position, % | 30.0 (13.0–60.0) | 10 |
| TLC, l | 3.3 (2.55–4.27) | 5 |
| TLC, % | 72.0 (52.9–90.0) | 9 |
| RV | 1.9 (1.12–2.0) | 7 |
| RV, % | 92.2 (35.0–104.0) | 8 |
| Ratio | 82.0 (69.0–98.4) | 9 |
| DLCO-SB, % | 67.9 (27.8–120.0) | 10 |
| IP max, % | 35.0 (32.8–50.6) | 11 |
| EP max, % | 76.0 (30.1–135.0) | 11 |
| SNIP, % | 30.5 (20–41) | 2 |
FEV1=forced expiratory volume in one second; FVC=forced vital capacity; TLC=total lung capacity; RV=residual volume; DLCO-SB=single-breath diffusing capacity of the lung for CO; IP max=decreased maximal inspiratory pressure; EP max=decreased maximal expiratory pressure; SNIP=sniff nasal inspiratory pressure; l/s=liters/second; l=liters.
Ref. 46.
Arterial blood gas analysis was performed at rest (FiO2: 21%). Median PaO2 was 64.5 (52.0–92.0)mmHg, median PaCO2 was 45.0 (30.0–65.2)mmHg, median O2 saturation was 92.5 (84.5–97.1)%, and median H2CO3 was 28.8 (23.5–36.0)meq/L.
Seven patients performed the 6-min walk (6MWDT) test using pulse oxymetry (SpO2) monitors. Six-min walk distance test (6MWDT) was defined as the longest distance possible in 6MWDT without encouragement. Overall, patients walked a mean distance of 334 (SD=129.8)m and had a mean fall of oxygen saturation of 7.1% [91.4% before and 84.3% after the test].
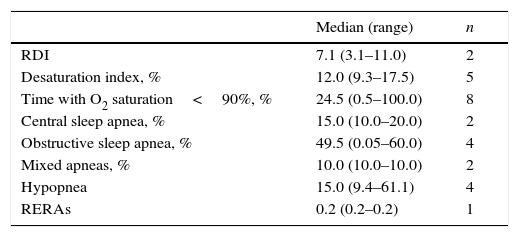
Polysomnography (PSG) testPSG results are shown in Table 6.
Polysomnography (PSG) test results.
| Median (range) | n | |
|---|---|---|
| RDI | 7.1 (3.1–11.0) | 2 |
| Desaturation index, % | 12.0 (9.3–17.5) | 5 |
| Time with O2 saturation<90%, % | 24.5 (0.5–100.0) | 8 |
| Central sleep apnea, % | 15.0 (10.0–20.0) | 2 |
| Obstructive sleep apnea, % | 49.5 (0.05–60.0) | 4 |
| Mixed apneas, % | 10.0 (10.0–10.0) | 2 |
| Hypopnea | 15.0 (9.4–61.1) | 4 |
| RERAs | 0.2 (0.2–0.2) | 1 |
RDI=respiratory disturbance index; RERA=respiratory effort-related arousal.
Electromyography was preformed in 6 patients and revealed normal results for 4 patients and altered results for the other 2.
Out of the 16 samples measured for dried blood spot (DBS) acid α-glucosidase, four tested positive for Pompe disease, a fifth patient evidenced a small reduction of acid α-glucosidase unconfirmed by serum GAA activity measurement, and the remaining 11 tested negative. The 2 remaining patients were tested for serum GAA activity (not DBS) and also tested negative for Pompe.
DiscussionThe late-onset form of Pompe disease has a heterogeneous presentation, mimicking other neuromuscular diseases, leading to diagnostic challenge. Clinical manifestations vary in terms of organ involvement, age at onset and severity, and symptoms are often unspecific and may remain mild for decades, so that neither the patient nor the doctor considered going further with diagnostic procedures. An early diagnosis could be relevant due to the chance of improving or at least stabilizing the course of disease through enzymatic replacement therapy (ERT).19,20
Although Pompe disease is a rare condition, it has been reported in a number of different ethnic populations, namely Caucasian, Taiwanese, Korean and Japanese.21–31 This study is the first to investigate the incidence of late-onset Pompe disease in Portugal, based on respiratory signs/symptoms. One clinical study had previously investigated and identified four cases of the juvenile form of the disease in Portuguese patients, raising awareness of the fact that Pompe disease should be suspected in progressive myopathies at any age, especially those involving limb-girdle and respiratory muscles and in small infants with cardiomyopathy.29
In the present study, 18 patients with a mean age of 60.8±11.1 years and diaphragmatic paralysis were identified and investigated for the late-form of Pompe disease. In Portugal, the diagnosis recommendations for late-onset Pompe disease suggest that patients with a progressive limb-girdle weakness, fatigue, cramps and muscle pain should be evaluated with creatinine kinase (CK) levels, electromyography, dynamic spirometry and muscle biopsy in inconclusive cases.16 Suspected cases and those in which muscle biopsy could not allow other diagnosis should be screened for lysosomal acid-α-glucosidase deficiency with dried blood spot (DBS), and the diagnosis should be confirmed by determination of lysosomal acid-α-glucosidase activity in a second sample and lysosomal acid-α-glucosidase gene sequencing. According to this, 5 of the 18 patients investigated evidenced absence or reduction of GAA activity in the DBS test.
Muscular weakness was a common symptom in these patients, with 61.5% of patients showing difficulty in getting up and 53.8% in sitting down, in keeping with other studies that have reported muscular weakness as the most common initial symptom in late-onset Pompe patients.8,32
Laboratory analysis revealed a mean CK value of 249.8 (±332.9; range: 24–1290)U/L, which is suggestive of Pompe disease, since CK values are typically elevated (>250U/L, equivalent to 1.5–15 times the upper limit of normal in adults) in all forms of this disease.
The exercise capacity (normally measured using the 6-min walk distance test [6MWDT]) and pulmonary function (forced vital capacity [FVC]) are outcome measures commonly used in studies of late-onset Pompe disease, by registering changes in the 6MWDT distance and in VC (measured as percentage change in predicted FVC [% predicted FVC]). In this study, patients walked a mean distance of 334±129.8m, which is in accordance with the 246–340m distance reported by other studies of late-onset Pompe patients.33–37 Although the 6MWDT is a valuable and widely used functional measure, it is associated with considerable inter- and intra-investigator variability, and does not elucidate the mechanisms behind the compromised physical function that it measures.38 Furthermore, the distance walked can be affected by factors such as patient motivation, age, sex, height, and weight as well as skeletal problems,39 which can affect gait and thereby influence the distance walked, as we noticed in this study.
FVC provides a simple measure of pulmonary function, and is a widely used outcome measure in studies of patients with neuromuscular disorders. Because diaphragm weakness often occurs early in the disease process and muscle weakness may not be evident until later in the disease course, respiratory evaluation is extremely important for patients who may have Pompe disease. Spirometric measurement of FVC in both the seated and supine positions is mandatory. A greater than 10% drop in FVC from the seated to the supine testing position is suggestive of diaphragm weakness and should raise the possibility of Pompe disease.40 Both measurements should be made regardless of whether respiratory symptoms are present. Because patients may have a normal seated FVC, the presence of diaphragm weakness can be missed if supine FVC is not measured.13,41 In this study, spirometry results revealed a 30% median drop in FVC when patients switched from the upright to the supine position, which is concordant with the diagnosis of diaphragm dysfunction.
Although not yet evaluated in late-onset Pompe disease, the sniff nasal inspiratory pressure (SNIP) test allows earlier detection of changes in respiratory muscle strength than FVC, and might be a useful measure to assess these patients.42
In this study, we could not find a statistically significant correlation between any of the functional test results, for either pulmonary function or muscle weakness, and Pompe disease, but diaphragm involvement was confirmed in the results for all the patients included.
The gold standard for diagnosis in Pompe disease is the determination of partial or complete deficiency of GAA enzyme activity in blood or fibroblasts. In this study, 18 patients with diaphragmatic paralysis were screened for Pompe disease, 2 through α-glucosidase serum activity measurements, 16 through the dried blood spot (DBS) α-glucosidase test, with positive results for 5 patients. In one patient Pompe disease was not confirmed by subsequent serum activity quantifications (which were normal). In three of the four positive patients the diagnosis of Pompe disease was established by gene sequencing, and all were recommended for enzymatic replacement therapy by the National Council for Lysosomal Storage Diseases. The fourth patient revealed only one previously identified mutation (D91N) related to a pseudo deficiency of GAA, and the diagnosis for Pompe disease remains unclear.
Due to the low prevalence and wide range of symptoms, Pompe disease is often unrecognized by physicians. Therefore, the development of a diagnostic methodology represents a valuable tool to enhance the recognition of Pompe disease by pulmonologists and other physicians. The American Association of Neuromuscular and Electrodiagnostic Medicine has developed an algorithm to aid in the diagnosis of Pompe disease,13 and other algorithms have been developed in recent years to allow for an earlier detection of late-onset Pompe disease cases.6,13,43–45 In patients presenting with diaphragm paralysis or respiratory symptoms suggestive of diaphragm muscle involvement, Pompe disease should be included in the differential diagnosis and patients should undergo further laboratory, electrodiagnostic and pulmonary evaluations, and imaging studies, as appropriate.
A simple DBS test can improve both the timing and accuracy of the diagnosis, which will lead to an improvement in the care of patients who have this progressively debilitating neuromuscular disease.
LimitationsWe were not able to establish a specific algorithm for the accurate diagnosis of late-onset Pompe disease in this population.
Another limitation of our study was that the non Pompe disease patients with diaphragm paralysis did not have a standardized neurological evaluation; these patients were followed by Pulmonologist only and it may be the case that a multidisciplinary evaluation is warranted for better characterization
Considering our primary objective, in this study we were able to diagnose Pompe disease in 3 of our pool of 18 patients (16.7%).
Although results from this study should be interpreted with caution due to the small sample size, we would like to highlight that Pompe's Disease is rare, with only 27 estimated cases undergoing ERT in an overall national population of approximately 10 million, and is our firm belief that Pompe should be suspected in all patients with diaphragmatic paralysis of unknown etiology and that DBS should always be performed in these patients.
Ethical responsibilitiesProtection of people and animalsThe authors state that no human or animal experiments have been performed for this investigation.
Confidentiality of the dataThe authors declare that no patient data appears in this article.
Right to privacy and written consentThe authors declare that no patient data appears in this article.
Conflicts of interestDr. Guimarães have received financial support for this investigation project, travel grants and honorariums for lecturing from Sanofi Genzyme® ref. GZ-2010-10243 – for DIPPER – DIaphragm Paralys is in Pompe Investigation.


