


ROS1 gene rearrangement is one of the druggable driver oncogenes that accounts for 1–2% of all non-small cell lung cancer (NSCLC), almost exclusively adenocarcinoma.1 Tumors harboring ROS1 gene rearrangements respond well to ROS1 tyrosine kinase inhibitors (TKIs), including crizotinib, ceritinib, entrectinib, and lorlatinib; however, resistance inevitably develops. Possible resistance mechanisms include genetic alteration of the drug target, activation of bypass signaling, or histological transformation to sarcomatoid carcinoma or small cell carcinoma.2 Here, we report a case of lung adenocarcinoma with CD74-ROS1 gene rearrangement that underwent sarcomatoid transformation during disease progression after crizotinib treatment. A tumor genome study also revealed a ROS1 F2004C mutation, and the patient was successfully treated with lorlatinib.
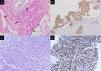
A 43-year-old non-smoker female was diagnosed with right upper lung adenocarcinoma (Fig. 1A) with an initial presentation of chronic cough for 3 months. The initial staging was cT4N3M1c with malignant pericardial effusion, spine metastasis, and solitary brain metastasis. ROS1 rearrangement was confirmed by both immunohistochemical stains (Fig. 1A) and fluorescence in situ hybridization (FISH). The patient was treated with cisplatin and pemetrexed for 1 cycle and then shifted to crizotinib due to the rapid progression of malignant pericardial and pleural effusion. The patient was kept on crizotinib for 10 months until an asymptomatic left-sided pleural mass was found during regular chest CT follow-up. A percutaneous sono-guided biopsy was performed at the pleural mass. Pathology showed pleomorphic spindle-shaped cells in solid sheets (Fig. 1C), with diffuse immunoreactivity for both cytokeratin (CK) and ROS1 (Fig. 1D).
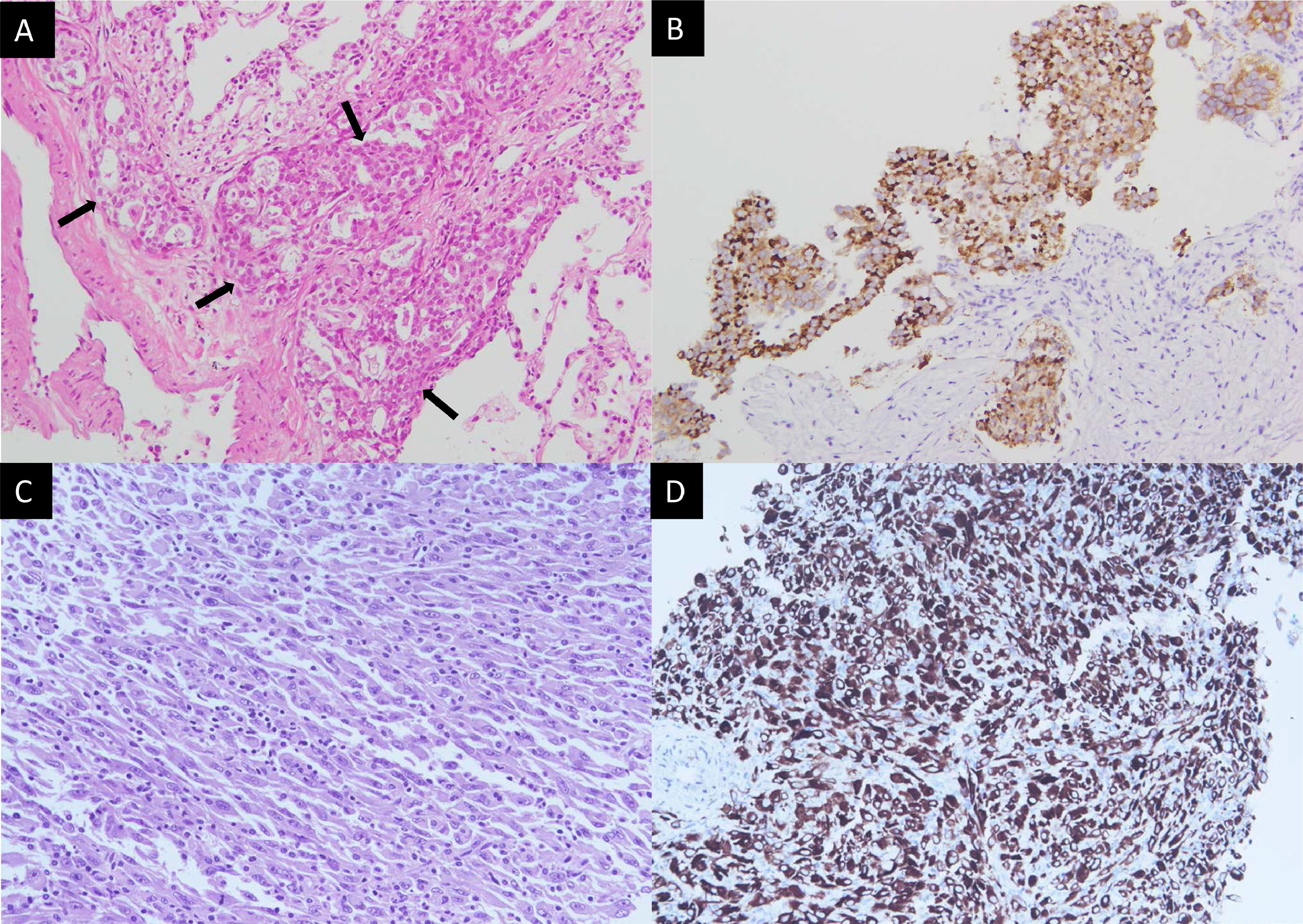
Pathology of the initial right upper lung tumor (A, B) and left pleural mass upon progression (C, D). (A) Hematoxylin and eosin stain showed round tumor cells (arrows) forming a glandular structure and solid nest. (B) Positive staining in ROS1 immunostaining. (C) Hematoxylin and eosin staining showed pleomorphic spindle-shaped tumor cells. (D) Tumor cells show positive staining in ROS1 immunostaining.
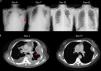
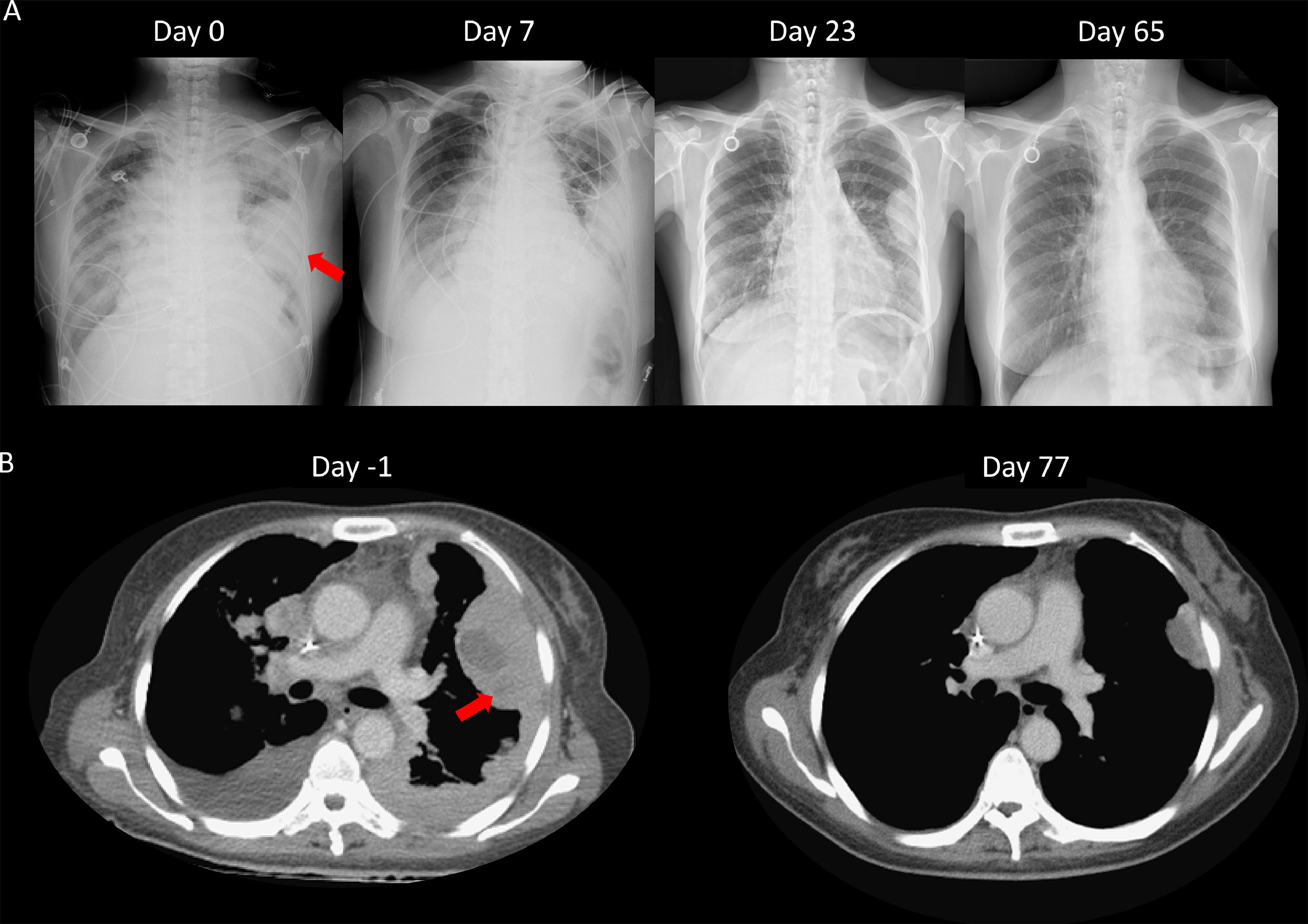
We speculated that the failure of first line chemotherapy treatment may be attributable to a larger tumor burden at the time; since chemotherapy often takes several weeks to achieve a clinical response, the patient received cisplatin and pemetrexed again. However, 3 weeks later, the patient was admitted to our emergency room (ER) due to severe dyspnea. Cardiac tamponade was diagnosed, and sudden collapse occurred with pulseless electrical activity noted during pericardiocentesis. The vital signs and consciousness gradually recovered after pericardiocentesis and intubation. Chest CT showed rapid progression of multiple pleural masses, malignant pericardial and pleural effusion, and lymphangitic carcinomatosis (Fig. 2B). After discussion with her family, lorlatinib 100mg was given via nasogastric tube. A pleural tumor specimen obtained previously was sent for next-generation sequencing (NGS) (Oncomine Focus Assay platform), and the result revealed a CD74-ROS1 fusion and ROS1 F2004C mutation. Extubation was performed 8 days after lorlatinib administration, and serial CXR (Fig. 2A) showed tumor regression, further confirmed by chest CT (Fig. 2B). The disease remained under control for 6 months.
Pulmonary sarcomatoid carcinoma is a rare histology type, accounting for only 0.1–0.4% of NSCLC.3 Lung adenocarcinoma with sarcomatoid transformation is even rarer. The optimal treatment modality for these patients is undecided. In a study from the Mayo Clinic involving the largest cohort of 127 patients with pulmonary sarcomatoid carcinoma, the response rate to palliative chemotherapy was only 8%, and the median overall survival was 7.7 months in stage IV disease.4 Recent studies have shown that pulmonary sarcomatoid carcinoma may harbor druggable oncogenes, such as MET-14 skipping, EGFR mutation, ALK translocation, and BRAF and ROS1 rearrangement.5 Several case reports have shown that such tumors may respond to relevant TKIs.
Crizotinib resistance mechanism is widely studied both in ALK and ROS1 rearrangement, including “on-target” crizotinib binding site secondary gene mutation, with ROS1 G2032R being most frequently identified. ROS1 F2004C/V was another acquired on-target mutation postulated to be treated by lorlatinib, another potent ALK/ROS1 TKI, using an in vitro model.6 Sarcomatoid transformation is another potential crizotinib resistance mechanism. Kobayashi et al. reported a case of ALK translocation adenocarcinoma in which disease progression occurred during crizotinib treatment following a biopsy that confirmed sarcomatoid transformation.7
In conclusion, Lorlatinib overcame both histological transformation and crizotinib binding site secondary mutation, suggesting that these sarcomatoid transformed cancer cells are still “addicted” to the ROS1 pathway. These findings highlight that histological transformation and other acquired mutations may coexist during disease progression; hence, comprehensive genetic testing is clinically valuable in determining resistance mechanisms to decide the next-line treatment.


