


Pulmonary alveolar proteinosis (PAP) is a rare respiratory disease and is pathogenetically classified into the following three types: autoimmune PAP, secondary PAP (sPAP), and congenital PAP.1 sPAP is not associated with anti-granulocyte macrophage colony-stimulating factor (GM-CSF) antibody and accounts for 5–10% of PAP cases. sPAP is primarily caused by hematological disorders; sPAP caused by autoimmune diseases is extremely rare2,3; therefore, clinicians may not always consider the possibility of sPAP when abnormal findings on chest radiography and/or computed tomography (CT) are seen in patients with autoimmune diseases. We report here, a case of sPAP in a patient with systemic lupus erythematosus (SLE).
A 70-year-old woman presented to our hospital with a 1-month history of exertional dyspnea. She was a known case of SLE and had a 37-year-history of treatment with corticosteroids and immunosuppressive agents. Her respiratory sounds were normal, and she had no fever or cough; her oxygen saturation was 94% in room air. Chest radiography and computed tomography revealed diffuse ground-glass opacities in the lung fields bilaterally (Fig. 1A–C). Serum level of Krebs von den Lungen-6, a marker of interstitial pneumonia, was elevated (6416 U/mL); β-d-glucan and cytomegalovirus antigenemia were absent. Non-infectious acute interstitial pneumonia was suspected, and a high-dose corticosteroid was administered; however, radiographic findings and symptoms did not improve. Furthermore, multiple nodules with cavitation appeared in the lower lobe of the left lung. Bronchoscopy was performed; the bronchoalveolar lavage fluid (BALF) was markedly cloudy (Fig. 1D), and showed deposition of acidophilic granular material and foamy macrophages on microscopy. Additionally, Nocardia was detected in the nodules of the left lower lobe. Serum anti-GM-CSF antibody was absent. Finally, she was diagnosed with sPAP associated with SLE, and pulmonary nocardiosis. Early treatment of nocardiosis was necessary; therefore, antibiotics were administered, while the corticosteroid dose was tapered. Unfortunately, pulmonary nocardiosis was refractory to treatment; furthermore, complications set in due to the cytomegalovirus pneumonia and non-tuberculous mycobacteriosis. Two months later, the patient died of exacerbating infections.
 Computed tomography image depicting diffuse ground-glass opacities in both lung fields. (D) Bronchoalveolar lavage fluid with a markedly cloudy appearance.")
In this case report, there were two important clinical observations. First, sPAP might develop in a patient with SLE. To the best of our knowledge, only one case of sPAP with SLE has been reported in the literature until date.4 According to previous reports, autoimmune diseases account for approximately 7% of the underlying causes of sPAP2,3; however, autoimmune diseases include Behcet’s disease, Wegener’s granulomatosis, microscopic polyangiitis, Sjogren’s syndrome, and dermatomyositis.2,3 There are few specific diseases; therefore, every autoimmune disease should be considered as a potential cause of sPAP.
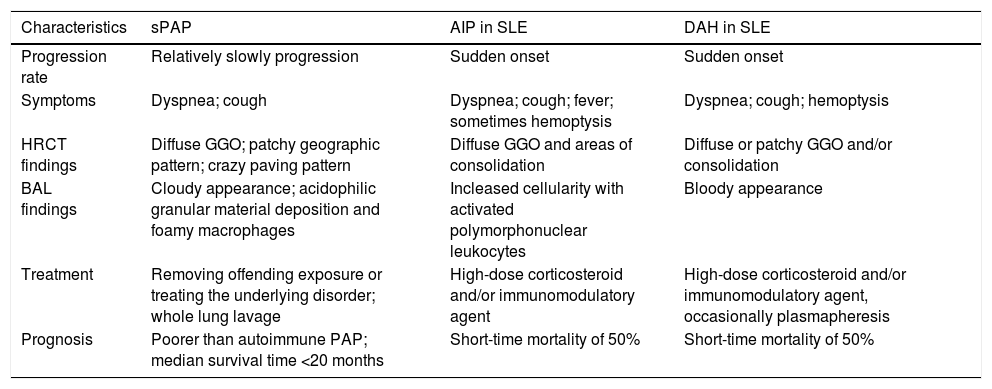
Second, chest CT findings of sPAP might be confused with those of acute interstitial pneumonia and pulmonary alveolar hemorrhage. In many cases of sPAP, such as the present one, chest CT shows ground-glass opacities in the lung fields bilaterally,5 which are also seen in acute interstitial pneumonia and diffuse alveolar hemorrhage, which are pulmonary disorders of SLE.6–8 The treatment of acute interstitial pneumonia and diffuse alveolar hemorrhage requires administration of high-dose corticosteroid and frequently an immunomodulatory agent as well. The treatment of sPAP needs to be quite different (Table 1) but as the mechanism underlying sPAP is still uncertain, there are no specific treatment indications for sPAP This may lead to high-dose corticosteroid and/or immunomodulatory agent being administered for pulmonary infections, as in case here presented. It is therefore important that in patients with SLE and diffuse pulmonary ground-glass opacities, clinicians should consider sPAP in addition to acute interstitial pneumonia and diffuse alveolar hemorrhage in the differential diagnosis.
Characteristics of secondary alveolar proteinosis compared to acute interstitial pneumonitis and diffuse alveolar hemorrhage in SLE.
| Characteristics | sPAP | AIP in SLE | DAH in SLE |
|---|---|---|---|
| Progression rate | Relatively slowly progression | Sudden onset | Sudden onset |
| Symptoms | Dyspnea; cough | Dyspnea; cough; fever; sometimes hemoptysis | Dyspnea; cough; hemoptysis |
| HRCT findings | Diffuse GGO; patchy geographic pattern; crazy paving pattern | Diffuse GGO and areas of consolidation | Diffuse or patchy GGO and/or consolidation |
| BAL findings | Cloudy appearance; acidophilic granular material deposition and foamy macrophages | Incleased cellularity with activated polymorphonuclear leukocytes | Bloody appearance |
| Treatment | Removing offending exposure or treating the underlying disorder; whole lung lavage | High-dose corticosteroid and/or immunomodulatory agent | High-dose corticosteroid and/or immunomodulatory agent, occasionally plasmapheresis |
| Prognosis | Poorer than autoimmune PAP; median survival time <20 months | Short-time mortality of 50% | Short-time mortality of 50% |
SLE, systemic lupus erythematosus; sPAP, secondary alveolar proteinosis; AIP, acute interstitial pneumonitis; DAH, diffuse alveolar hemorrhage; HRCT, high resolution computed tomography; GGO, ground-glass opacities; BAL, bronchoalveolar lavage.
sPAP has a poor prognosis compared to autoimmune PAP.2 Acute interstitial pneumonia and diffuse alveolar hemorrhage associated with SLE have sudden onset with severe symptoms, while the progression of sPAP is relatively slow and symptoms are relatively mild. Regular and close follow-up on chest radiography is necessary for early detection of sPAP in patients with SLE. For early initiation of appropriate treatment, awareness of the possibility of sPAP in patients with ground-glass opacities in both lung fields is important.
Author contributionsStudy conception and design: Masahiro Yamasaki.
Data acquisition: Masahiro Yamasaki, Kazuma Kawamoto, Shota Nakano, Masaya Taniwaki, Naoko Matsumoto, Shinji Nabeshima.
Data analysis and interpretation: Masahiro Yamasaki, Kazuma Kawamoto, Shota Nakano, Noboru Hattori.
Drafting of the manuscript: Masahiro Yamasaki.
Critical revision of the manuscript for important intellectual content: Noboru Hattori.
All authors had access to the data and played a role in writing this manuscript.
Conflict of interestNone declared.
We thank Editage (www.editage.jp) for English language editing.


