


As there are few studies on the impact of respiratory and functional status on the quality of life domains in adults with cystic fibrosis, this study aimed to evaluate the association between respiratory function, functional capacity and quality of life in these subjects.
MethodsThis is a cross-sectional study, where adults with clinical and laboratorial diagnoses of CF fibrosis underwent pulmonary function tests, the six-minute walk distance test (6MWT) and responded to the Cystic Fibrosis Questionnaire-Revised (CFQ-R). Descriptive statistics was used to summarize the findings. The associations were tested by means of Pearson's or Spearman tests, and the significance level was set at 5%.
ResultsThe 21 patients who completed the study presented with reduced quality of life in all CFQ-R domains, obstructive pulmonary disease and reduced 6MWT distance. The following associations were found between pulmonary function and CFQ-R domains: forced vital capacity – FVC (%) and treatment burden and digestive symptoms (r = −0.433, p < 0.05; r = −0.443, p < 0.05, respectively), forced expiratory volume in one second – FVC ratio – FEV1/FVC (%) and physical functioning, social and respiratory symptoms (r = 0.5, p < 0.05; r = 0.58, p < 0.01; r = 0.45, p < 0.05, respectively), residual volume (%) and physical functioning (r = 0.49, p < 0.05), airways’ resistance – Raw and physical functioning and emotional functioning (r = −0.44, p < 0.05; r = −0,46, p < 0.05, respectively), carbon monoxide diffusing capacity (%pred) and physical functioning (r = −0,51; p < 0.05).
ConclusionAdults with CF have reduced quality of life, which in part is associated with the severity of their lung function.
Cystic fibrosis (CF) is a hereditary, autosomal recessive multisystem disease characterized by obstructive pulmonary disease, pancreatic insufficiency, malnutrition and high levels of electrolytes in sweat.1 It is caused by mutation in the CFTR protein (Cystic Fibrosis Transmembrane Conductance Regulator) located on the long arm of chromosome 7,2 and has a prevalence of 1 in every 2500 live births.3
For more than 40 years, CF was fatal in infancy, and although it remains incurable, great scientific advances have resulted in increased survival from 16 to 35 years in 2004.3 In 2008, the United Kingdom and the United States estimated the average survival of these individuals in 38.8 and 37.4 years, respectively. It is estimated that newborns with CF in the twenty-first century will survive until over 50 years of age.4
The respiratory impairment is progressive, with variable intensity and occurs in 95% of adults with CF, the lung injury being the most important determinant for prognosis.5 In the airways, the defect in CFTR protein reduces the chloride ions secretion and promotes a higher sodium and water absorption, resulting in dehydrated, thick and viscous respiratory mucus. This mucus accumulates in the lungs predisposing to repeated lung infections.6 Thus, the ventilatory defect in CF is essentially obstructive. Only in the final phase a restrictive component arises due to pulmonary fibrosis, which leads to reduced lung volumes.7
Measures of functional capacity have been increasingly used in the follow-up of adults with CF in order to monitor disease progression and treatment effects, and to assess the prognosis of the individuals and their ability to exercise.3
Besides the functional and pulmonary impairments, the impact of disease on quality of life of adults with CF is influenced by several factors, including the dysfunction of multiple organs and need for complex treatments,8 as well as psychological and social aspects.9 Therefore, the quality of life contributes to the assessment of overall treatment efficiency and helps adjust the multidisciplinary team approach.1 This study aimed at evaluating the association between pulmonary function, functional capacity and quality of life in adults with cystic fibrosis.
MethodsSubjectsThis is a cross-sectional study, including subjects recruited from the Policlínica Piquet Carneiro – Universidade do Estado do Rio de Janeiro (UERJ), Brazil. Between September 2011 and August 2012, adults with CF (aged 18 years or older) were included if they had clinical and laboratorial diagnosis of CF (sweat test and/or deoxyribonucleic acid – DNA mutation analysis). Exclusion criteria were: history of disease exacerbation within the last four weeks preceding the study or inability to perform the pulmonary function and exercise tests. The study was approved by the Research Ethics Committee of the Centro Universitário Augusto Motta (UNISUAM), Rio de Janeiro, RJ, Brazil under protocol number 015/11 and all participants signed an informed consent.
MeasurementsPulmonary function, six-minute walk distance tests (6MWT) and the Cystic Fibrosis Questionnaire-Revised (CFQ -R) domains were recorded at the Laboratório de Função Pulmonar do Departamento de Pneumologia do Hospital Universitário Pedro Ernesto (HUPE/UERJ). The pulmonary function screening was performed in accordance to the recommendations of the American Thoracic Society (1995)10 and the Brazilian Thoracic Society (2002),11 and used the Collins Plus Pulmonary Function Testing Systems (Warren E. Collins, Inc., Braintree, MA, USA). The predicted values were set according to the equations described by Pereira (spirometry),12 Neder (pulmonary volumes),13 Neder (diffusion)14 and Neder (maximal respiratory pressures).15 The 6MWT was performed in accordance with the American Thoracic Society recommendations,16 and the predicted reference equations of Soares were used for the analysis.17
We used the Brazilian validated version of the CFQ-R 14,8 which is appropriate to assessing patients over 14 years and adults. The questionnaire has 50 questions, divided into 12 domains. Its values range from 0 to 100 points and the higher the score, the better quality of life.
Statistical analysisAll statistical analyses were performed with SigmaStat 3.5 (Systat Software, San Jose, CA, USA). Descriptive statistics included means and standard deviations for continuous measures and counts and percentages for categorical measures. Data distribution was tested using the Shapiro–Wilk test, and then Pearson's or Spearman tests were used for correlations. Statistical significance was considered for p < 0.05.
ResultsOf the 48 participants eligible for evaluation, 21 completed the study (Figure 1). Anthropometric data, pulmonary function and CFQ-R domains are shown in Table 1. Associations between pulmonary function and CFQ-R domains are presented in Table 2.
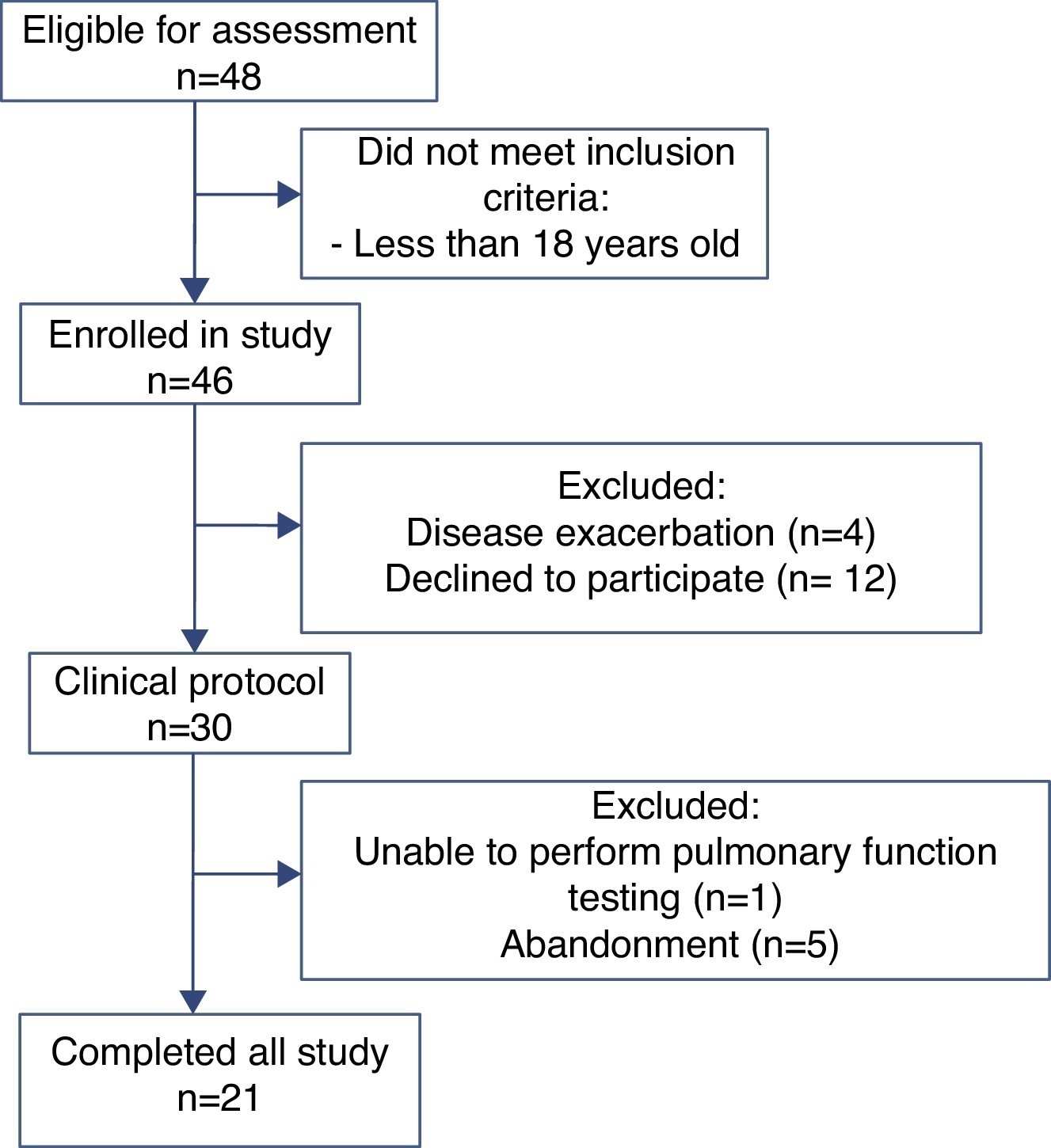
Figure 1. Flow chart showing the different stages of the recruitment process.
Table 1. Anthropometric data, pulmonary function and Cystic Fibrosis Questionnaire-Revised domains.
| Variables | Values |
| Demographic characteristics | |
| Sex (male) | 12 (57) |
| Age (years) | 25.5 (±6) |
| Height (cm) | 166 (±0.1) |
| Weight (kg) | 58.2 (±14) |
| Prolife of chronic pulmonary infection | |
| No chronic pulmonary infection, n (%) | 4 (19) |
| Chronic P. aeruginosa infection, n (%) | 13 (62) |
| Chronic Burkholderia cepacia complex, n (%) | 4 (19) |
| Pulmonary function parameters | |
| FEV1 (% predicted) | 57 (±26) |
| FVC (% predicted) | 75 (±25) |
| FEV1/FVC (%) | 64 (±15) |
| TLC (% predicted) | 106.7 (±23) |
| RV (% predicted) | 195 (±90) |
| RV/TLC (%) | 176 (±64) |
| Raw (cmH2O/L/s) | 2.5 (±2.0) |
| DLco (%predicted) | 76 (±42) |
| CFQ-R scores | |
| Physical functioning | 67.7 (±17.2) |
| Role | 74.6 (±16.8) |
| Social | 62.4 (±22.3) |
| Body image | 69.3 (±30.4) |
| Respiratory symptoms | 68.7 (±19.2) |
| Vitality | 67.5(±18.2) |
| Emotional functioning | 75.2 (±15.1) |
| Eating disturbances | 88.4 (±18.4) |
| Treatment burden | 64 (±20.5) |
| Health perceptions | 57.7 (±19.8) |
| Weight | 55.6 (±37) |
| Digestive symptoms | 81 (±18) |
Results expressed as mean (SD) or number (%). FEV1: forced expiratory volume in one second; FVC: forced vital capacity; TLC: total lung capacity; RV: residual volume; Raw: airway resistance; DLco: carbon monoxide diffusing capacity; CFQ-R: Cystic Fibrosis Questionnaire Revised.
Table 2. Pearson correlation coefficients between pulmonary function and Cystic Fibrosis Questionnaire-Revised domains in adults with cystic fibrosis.
| Variables | Physical Functioning | Social | Respiratory Symptoms | Emotional Functioning | Treatment Burden | Digestive Symptoms |
| FVC (%) | – | – | – | – | −0.433 * | −0.443 * |
| FEV1/FVC (%) | 0.500 * | 0.582 ** | 0.446 * | – | – | – |
| RV (%) | −0.485 * | – | – | – | – | – |
| Rwa cmH2O/L/s | −0.437 * | – | – | 0.461 * | – | – |
| DLco (%) | 0.516 * | – | – | – | – | – |
FEV1: forced expiratory volume in one second; FVC: forced vital capacity; TLC: total lung capacity; RV: residual volume; Raw: airways’ resistance; DLco: carbon monoxide diffusing capacity; PImax: maximal inspiratory pressure; PEmax: maximal expiratory pressure.
* p < 0.05.
** p < 0.01.Only the correlation coefficients from statistically significant associations are shown.
There were associations between the 6MWT distance and the following pulmonary function variables: forced expiratory volume in one second – FEV1 (r = 0.618, p = 0.003), forced vital capacity – FVC (r = 0.632, p = 0.002), carbon monoxide diffusing capacity – DLCO (r = 0.506, p = 0.02), residual volume (RV) – total lung capacity (TLC) ratio – RV/TLC (r = −0.496, p = 0.02) and airway resistance – Raw (r = −0.741, p = 0.0001). There were no associations between the 6MWT distance and the CFQ scores.
DiscussionOur findings showed that the severity of pulmonary disease is associated with quality of life and functional capacity in adult patients with CF. These associations were found for some CFQ-R domains, even those not related to pulmonary function. It confirms the multisystem profile of the disease, and suggests that the pulmonary impairments may reflect the overall progression and severity of the disease.
Among the main pathophysiological findings in CF, lung function severity is considered an important mortality and morbidity marker.3, 18, 19 Thus, a more severely impaired lung function is commonly associated with a worse metabolic, nutritional and digestive function, which in turn, may explain the correlations found in our study between the pulmonary function variables, CFQ-R domains and the 6MWT results. The worsening of lung function associated with reduced exercise tolerance starts a vicious cycle where the patient abstains from physical activity, resulting in progressive deconditioning and increased dyspnoea. Although this physical limitation compromises daily life activities, impacting negatively on the quality of life in adults with CF,20 we found no studies that have assessed the correlation between the functional capacity (6MWT) and the quality of life in adults with cystic fibrosis. In children and adolescents, Donadio et al. found an association between the physical domain of the CFQ-R and the 6MWT.21 In our study, maybe because of the small sample size, there was no association between the CFQ scores and the 6MWT. Therefore, our results suggest that the pulmonary function may reflect the overall disease severity better than the functional capacity, and that the exercise performance is not necessarily the foremost factor influencing the quality of life in such subjects. Moreover, some studies observed no differences in the CFQ-R physical domain after a supervised exercise program.22, 23 Maybe this domain does not reliably reflect the exercise performance of such patients, which may have influenced the absence of association between the exercise capacity and quality of life in our study.
In addition to the pulmonary impairments, other systemic characteristics of the disease also affect the quality of life in adults with CF. In fact our study showed that six of the 12 CFQ-R domains showed no correlation with lung function. Given the complexity of the pathophysiology of the disease, it is expected that lung function alone cannot show the impact of disease on quality of life in adults with CF. Gastrointestinal manifestations are associated with frequent episodes of abdominal pain, constipation, loss of appetite, nausea, weight loss and compromised nutritional status. These changes may limit CF individuals in their daily activities and therefore, compromise the quality of life.24, 25
In a similar way to the study of Abbott et al.,26 our results indicated that the pulmonary function may be a contributory factor for the psychosocial profile in adults with CF. It is likely that the respiratory symptoms such as cough, dyspnea and chest pain contribute to social isolation. Consequently, further changes may arise, such as anxiety, depression and even sleep disorder. These factors may negatively affect the quality of life in adults with CF.9, 24, 27, 28, 26
As indicated by the “Treatment burden” domain, the time available for the daily treatment may also impact on the quality of life of these subjects. The time for activities dedicated to the treatment is considered high. On average, adults with CF require 108 min to complete the daily routine treatment. The respiratory care requires more time, comprising, on average, 41 min for nebulization and 29 min for airway clearance techniques.29 Besides the longer time, the complexity of treatment is also considered higher in adults compared with children and adolescents with cystic fibrosis.30 In adults, the severity and exacerbations of lung function are more frequent and reflect the morbidity,31 making the treatment burden scores higher and, thus the perception of overall quality of life worse. This is in accordance with the score below 70% found for many CFQ-R domains in our study (physical, social, body image, respiratory, vitality and treatment burden). Conversely, Donadio et al.21 assessed the pulmonary function and quality of life in a sample of children and adolescents with CF and found normal scores for most domains.
The strength of this study is that it adds new knowledge on the relationship between quality of life and lung function in adults with CF. Currently, many individuals with CF live beyond their 50 s, and the number of adults with CF has grown remarkably; this makes the knowledge of this new population of individuals highly relevant given the new treatment perspectives. One limitation of our study is the small sample size. From the 48 initially screened patients for the study, many dropped out of the protocol; non-attendance for assessment visits being the most common reason (12 declined to participate, and 5 abandoned). To some extent, this is an expected result, since adults with CF are often demotivated, as demonstrated by the “Social” and “Emotional functioning” scores observed in our results. Another limitation is that it is a cross-sectional design, which prevents the evaluation of the long-term consequences of the disease. Despite these limitations, our findings indicate that, not only pulmonary function and functional capacity, but also the quality of life is an important issue in the follow-up of adult patients with CF. The different CFQ domains provide a comprehensive understanding on how the disease impacts on patients’ daily life, which, in turn, can support more individualized and efficient approaches. Moreover, few authors have assessed the association between pulmonary function, functional capacity and quality of life. Therefore, our results suggest that the systemic manifestations, the functional capacity and the pulmonary function severity influence the quality of life in adults with CF.
ConclusionAdults with CF have reduced quality of life and functional capacity, which in part are associated with the severity of their lung function. These findings can support a better understanding of the overall disease burden and help the clinical follow-up of these patients.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
Acknowledgement
This research was supported by the Research Support Foundation of the State of Rio de Janeiro; Brazil (FAPERJ).
Received 19 June 2014
Accepted 21 October 2014
Corresponding author. fguimaraes_pg@yahoo.com.br


