






A1Antitrypsin deficiency (AATD) pathogenic mutations are expanding beyond the PI*Z and PI*S to a multitude of rare variants.
Aimto investigate genotype and clinical profile of Greeks with AATD.
MethodsSymptomatic adult-patients with early-emphysema defined by fixed airway obstruction and computerized-tomography scan and lower than normal serum AAT levels were enrolled from reference centers all over Greece. Samples were analyzed in the AAT Laboratory, University of Marburg-Germany.
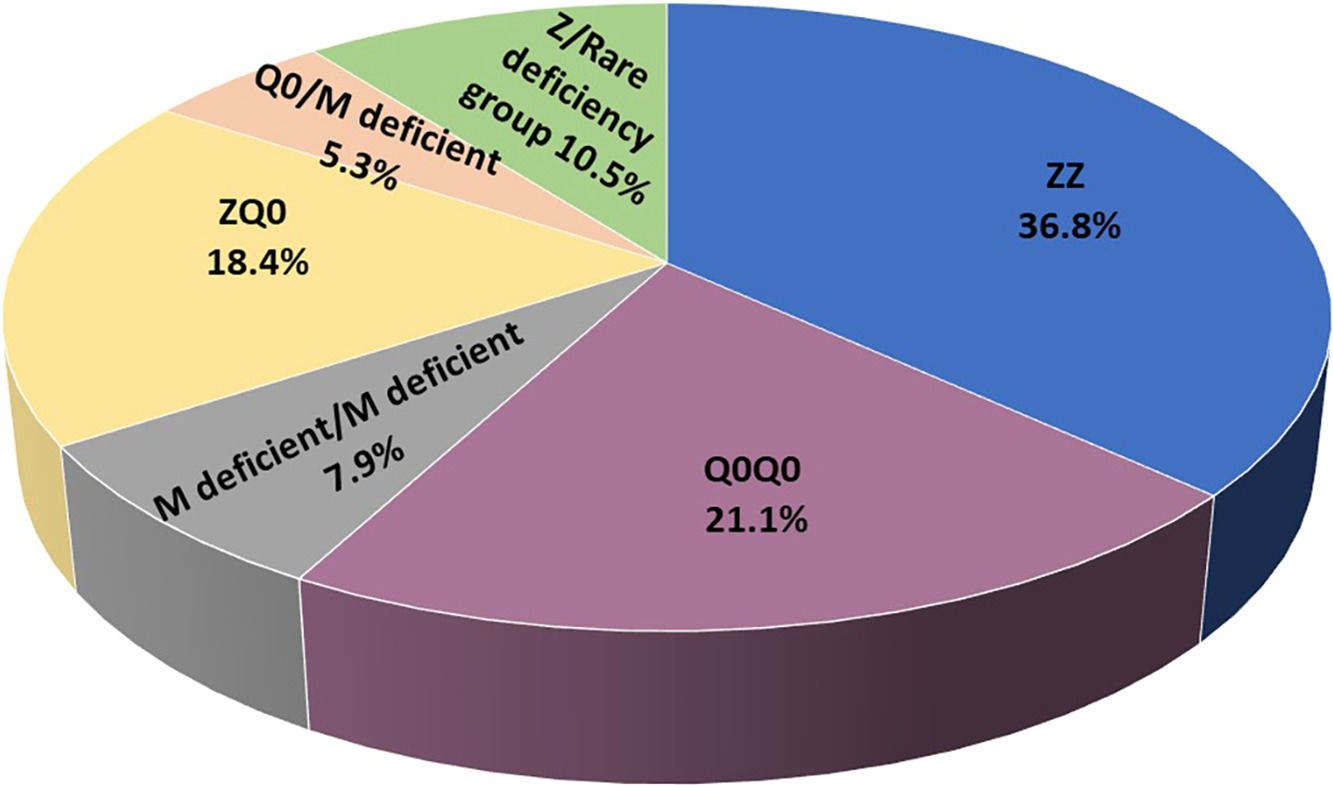
ResultsIncluded are 45 adults, 38 homozygous or compound heterozygous for pathogenic variants and 7 heterozygous. Homozygous were 57.9% male, 65.8% ever-smokers, median (IQR) age 49.0(42.5–58.5) years, AAT-levels 0.20(0.08–0.26) g/L, FEV1(%predicted) 41.5(28.8–64.5). PI*Z, PI*Q0, and rare deficient allele's frequency was 51.3%, 32.9%,15.8%, respectively. PI*ZZ genotype was 36.8%, PI*Q0Q0 21.1%, PI*MdeficientMdeficient 7.9%, PI*ZQ0 18.4%, PI*Q0Mdeficient 5.3% and PI*Zrare-deficient 10.5%. Genotyping by Luminex detected: p.(Pro393Leu) associated with MHeerlen (M1Ala/M1Val); p.(Leu65Pro) with MProcida; p.(Lys241Ter) with Q0Bellingham; p.(Leu377Phefs*24) with Q0Mattawa (M1Val) and Q0Ourem (M3); p.(Phe76del) with MMalton (M2), MPalermo (M1Val), MNichinan (V) and Q0LaPalma (S); p.(Asp280Val) with PLowell (M1Val); PDuarte (M4), YBarcelona (p.Pro39His). Gene-sequencing (46.7%) detected Q0GraniteFalls, Q0Saint-Etienne, Q0Amersfoort(M1Ala), MWürzburg, NHartfordcity and one novel-variant (c.1A>G) named Q0Attikon.Heterozygous included PI*MQ0Amersfoort(M1Ala), PI*MMProcida, PI*Mp.(Asp280Val), PI*MOFeyzin. AAT-levels were significantly different between genotypes (p = 0.002).
ConclusionGenotyping AATD in Greece, a multiplicity of rare variants and a diversity of rare combinations, including unique ones were observed in two thirds of patients, expanding knowledge regarding European geographical trend in rare variants. Gene sequencing was necessary for genetic diagnosis. In the future the detection of rare genotypes may add to personalize preventive and therapeutic measures.
A1Antitrypsin (AAT) which is mainly produced by hepatocytes, acts as major protease inhibitor in the serum and targets preferentially excessive human neutrophil elastase.1 Normally its plasma levels range from 0.9 g/L to 2 g/L in the absence of an acute phase response. AAT deficiency (AATD) is characterized by a significant decrease of AAT plasma levels usually below the “protective level” of 0.57 g/L.2,3 Low or absent plasma levels and/or dysfunctional AAT molecules increase risk to develop pulmonary emphysema, less commonly liver disease and rarely other systemic manifestations.4–7 Cigarette smoking or equivalent is considered the major additional risk factor in these patients.1,2 AATD is one of most common Mendelian disorders and PI*Z and PI*S variants account for the majority of cases worldwide.8,9 However, the genetic repertoire of AATD pathogenic mutations is constantly expanding far beyond the PI*Z and PI*S variants to a multitude of rare alleles such as the deficient or dysfunctional ones leading to the production of misfolded AAT protein such as the M, N, P, O deficient and the null (Q0) variants without any production of AAT protein.10,11The prevalence of rare genotypes other than PI*ZZ and PI*SZ recorded so far ranges from 1.1% in countries like France to 20.4% in countries like Italy.12–16 Their epidemiology and geographic distribution, pathogenetic role and clinical expression need further investigation. This study aimed to investigate for the first ever time in Greece genotype and clinical profile characteristics of AAT deficiency patients.
Patients and methodsIn this retrospective collaborative Greek study, we assessed the genotype and clinical profile of AAT deficiency patients followed-up in 12 hospital centers all over Greece a country of 10.400.000 people [https://www.statistics.gr/press-kit_census_results_2021] in an effort to capture the entire population. For that purpose, after authorization by the Medical Ethics Committee of General University Hospital “Attikon”, Athens, Greece, a questionnaire was addressed to all Tertiary University Hospitals as well as Referral Centers of the National Health System (NHS) and dedicated private clinics to enroll symptomatic adult patients with early emphysema defined by fixed airway obstruction and computerized tomography scan findings and lower than normal serum AAT levels documented by two consecutive measurements in the absence of an acute reaction condition verified by normal CRP levels. The overlap presence of bronchiectasis, bronchial asthma and liver disease was also reported in addition to routinely collected epidemiologic, clinical and functional data. This case-finding program is still ongoing. The data of the present study correspond to the time period from September 2018 to October 2021. Normal serum AAT levels were considered at 0.9–2 g/L by nephelometry by local laboratory standards. Following genetic results, analysis considers adults as homozygous or compound heterozygous for pathogenic variants. All adult patients included in the study fulfilled the criteria of the European Respiratory Society for the diagnosis and treatment of pulmonary disease in AATD.3The AATD genetic analysis was performed at the German AAT Laboratory at the University of Marburg (MV, CFV, TG). For that analysis dried blood spot (DBS) samples were tested using the Progenika AAT genotyping kit (Progenika Biopharma, S, A, Derio, Spain) which can simultaneously identify and genotype 14 deficiency variants of the SERPINA1 gene based on Luminex xMAP-Technology (Luminex, Austin, TX, USA). For samples in which we suspected the presence of other mutations, due to low serum AAT levels, isoelectric focusing (IEF) was performed.17,18 If the results from Luminex technology and IEF tests did not correlate or an indication of a rare mutation existed, gene sequencing (Next Generation Sequencing) was done. For sequencing the DBS samples were shipped to a reference laboratory [Progenika Biopharma, Spain]. We have described the laboratory methods in detail elsewhere.17 Epidemiological, clinical, laboratory and functional data were collected in a standardized manner. Death data were recorded until October 2021, except for cases lost to follow-up. Rare and ultra-rare mutations were discussed systematically with experts from the German AAT Laboratory at the University of Marburg (MV, CFV, TG) in collaboration, when necessary, with the expert Center for Diagnosis of Inherited Alpha1-antitrypsin Deficiency at the University of Pavia on web meetings (IF, SO). Selected patients were discussed monthly in a web-based multidisciplinary meeting dedicated to AATD (www.respifil.fr) for consultation (MB, CL, VC, JFM). All patients signed written informed consent and the study was approved by the Medical Ethics Committee of General University Hospital “Attikon”, Athens, Greece (ΕΒΔ 633/24–09–2018).
Statistical analysisNormality of the distributions was checked with Kolmogorov-Smirnov test. Categorical variables are presented as n (%), whereas numerical variables are presented as median (interquartile ranges) since the distribution of data was skewed. Comparisons between AAT levels in the different groups were performed using Kruskal Wallis test. Statistical significance was established at the level of p ≤ 0.05. Data were analyzed using SPSS 17.0 for Windows (SPSS Inc., Chicago, IL, USA) and graphs were created using Graph Pad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
ResultsIncluded are 45 adults, 38 homozygous or compound heterozygous for pathogenic variants and 7 heterozygous. The former was 57.9% male, 65.8% ever-smokers, diagnosed at a median age (IQR) of 49.0 (42.5–58.5) years, AAT levels of 0.20(0.08–0.26) g/L, FEV1(% predicted), FEV1/FVC%, FEF25–75% and DLCO (% predicted) of 41.5 (28.8–64.5), 49.6 (38.2–61.1), 14 (9.6–28.5) and 45.8 (31.7–58.3), respectively. Overall, 26.3% of patients presented overlapping emphysema and bronchiectasis and 2.6% bronchial asthma. Only 2.6% presented with liver disease. All patients were treated with a combination of LABA/LAMA bronchodilators in the addition of inhaled corticosteroids in 50%. Augmentation therapy was provided in 26 (68.4%) patients although 30 of them (78.9%) fulfilled all 3 criteria for treatment (age <70 years old, FEV1< 70% and AAT levels less than 0.57 g/L).19 No vasculitis was reported in the present cohort analysis. PI*Z, PI*Q0, PI*Mdeficient and PI*N allele's frequency was 51.3%, 32.9%, 14.5% and 1.31%, respectively. PI*ZZ genotype was found in 36.8% of patients, PI*Q0Q0 in 21.1%, PI*MdeficientMdeficient in 7.9%, PI*ZQ0 in 18.4%, PI*Q0Mdeficient in 5.3% and PI*Zrare-deficient in 10.5% (Table 1, Fig. 1).
Epidemiological, clinical and laboratory data of adults homozygous or compound heterozygous for pathogenic variants.
Values are presented as median (IQR) unless otherwise indicated; IQR= interquartile range; p-values <0.05 were considered statistically significant, M=male; COPD= chronic obstructive pulmonary diseases; FEV1= Forced expiratory volume in one second FVC= forced vital capacity; FEF25–50= forced expiratory flow at 25–75% of forced vital capacity or forced mid-expiratory flow; DLCO= diffusing capacity of the lung for carbon monoxide; all 3 criteria: age <70 years old, FEV1< 70% and AAT levels less than 0.57 g/L.
Distribution of patients all over Greece and in Cyprus is shown in Fig. 2. Using genotyping by Luminex the following mutations were detected: p.(Pro393Leu) which is associated with MHeerlen (M1Ala/M1Val); p.(Leu65Pro) which is associated with MProcida; p.(Lys241Ter) which is associated with Q0Bellingham; p.(Leu377Phefs*24) which is associated with Q0Mattawa (M1 Val) and Q0Ourem (M3); p.(Phe76del) which is associated with MMalton (M2), MPalermo (M1Val), MNichinan (V) and Q0LaPalma (S); p.(Asp280Val) which is associated with PLowell (M1Val) ; PDuarte (M4), YBarcelona (p.Pro39His) (Table 2). However, this method does not detect the background, as is the case with sequencing. Therefore, in the following text the mutations are written as follows:
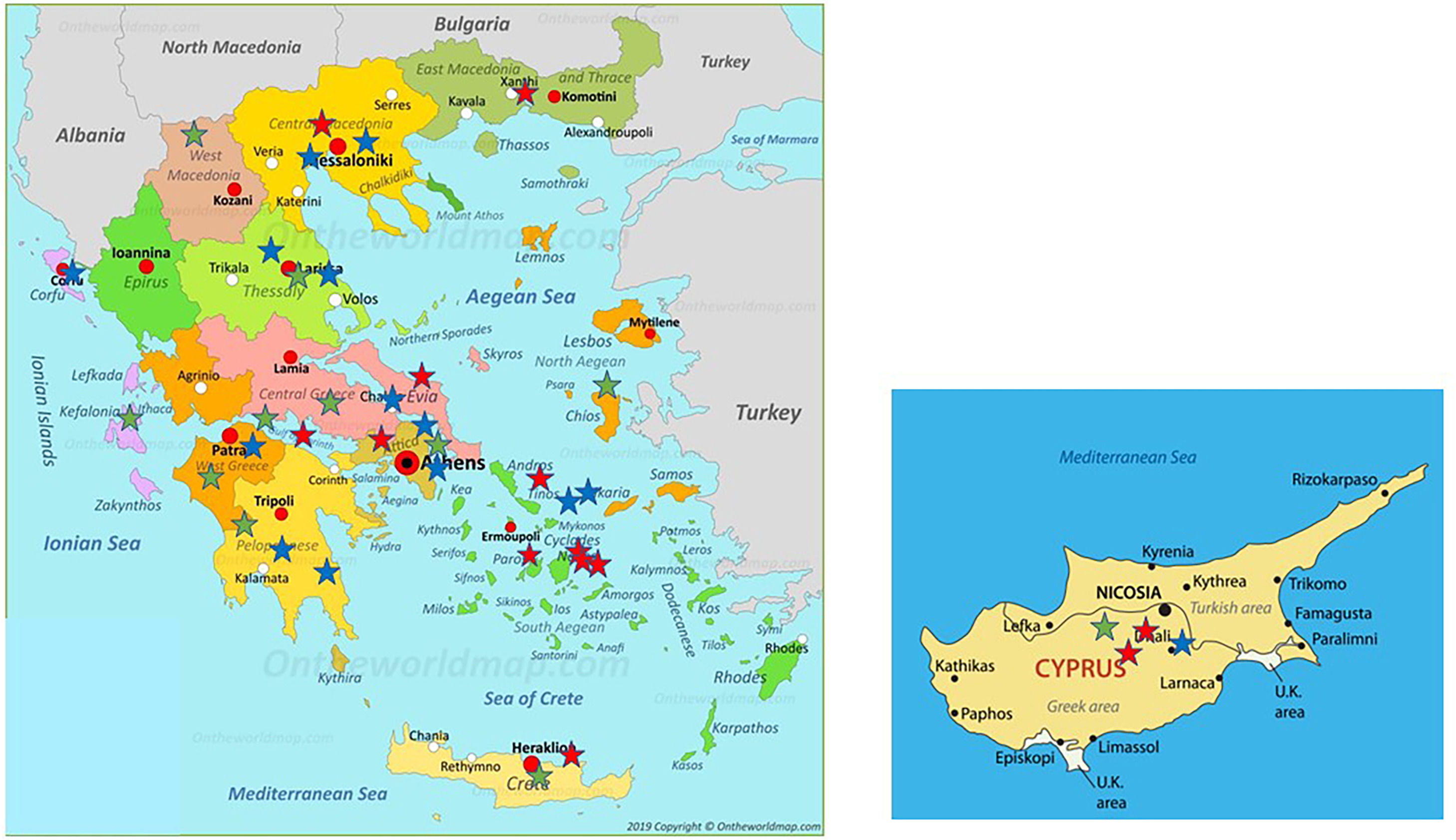
Distribution of the 38 patients depicted as stars of different colors in their area of origin on the map of Greece and Cyprus. Blue stars correspond to PI*ZZ patients, red stars to any combination homozygous or compound heterozygous including PI*Q0 or/and rare-deficient variants and green stars correspond to the combination of PI* Z with any PI*Q0 or rare-deficient variant.
Biological characteristics of the alleles of the study population.
| SERPINA1 mutation | Marker ID | Allele name (Molecular Background) | DNA Sequence | Intron/Exon | Mechanism | Mutation effect on the protein | Refs. |
|---|---|---|---|---|---|---|---|
| p.(Thr62Cys) | OFeyzin | c.185A>G | Exon 4 | Missense mutation | unknown | http://www.a1at-variantdatabase.com/detail-page-display-260.html | |
| p.(Leu65Pro) | rs28931569 | MProcida | c.194T>C | Exon 2 | Missense mutation | Protein deficiency | 28 |
| p.(Phe76del) | rs775982338 | MMalton (M2)MPalermo (M1 Val)MNichinan (V)Q0la palma (S) | c.227_229delTCTdel | Exon 2 | In-frame-deletionSingle amino acid deletion at position 76 | Protein deficiency | 30,36–38 |
| p.(Asp280Val) | rs121912714 | PLowell (M1Val)PDuarte (M4)YBarcelona (p.Pro39His) | c.839A>T | Exon 5 | Missense mutation | Protein deficiency | 8 |
| p.(Tyr184Ter) | rs199422210 | Q0Amersfoort (M1Ala)Q0Bredevoor (Unknown) | c.552C>G | Exon 2 | Nonsense mutationThis sequence change creates a premature translational stop signal (p.Tyr184Ter) in the SERPINA1 gene. It is expected to result in an absent or disrupted protein product | Protein absence | 8,27 |
| p.(Tyr184Ter) | rs267606950 | Q0Granite Falls | c.552del | Exon 2 | Frameshift mutationThis frameshift results in a premature stop signal (p.Tyr184Ter) in the SERPINA1 gene. It is expected to result in an absent or disrupted protein product | Protein absence | 29 |
| p.(Lys187Ter) | Q0Saint-Etienne | c.559A>T | Exon 2 | Nonsense mutationThis sequence change creates a premature translational stop signal (p.Lys187Ter) in the SERPINA1 gene. It is expected to result in an absent or disrupted protein product. | Protein absence | 26 | |
| p.(Lys241Ter) | rs199422211 | Q0Bellingham | c.721A>T | Exon 3 | Nonsense mutationThis sequence change creates a premature translational stop signal (p.Lys241Ter) in the SERPINA1 gene. It is expected to result in an absent or disrupted protein product. | Protein absence | 31 |
| p.(Thr292Ile) | rs745624643 | NHartford City | c.875C>T | Exon 5 | Missense mutation | Protein deficiency | 32 |
| p.(Leu377Phefs*24) | rs763023697 | Q0Mattawa (M1 Val)Q0Ourem (M3) | c.1130dup | Exon 5 | Frameshift mutationAn insertion of 1 nucleotide at position 353 of the protein resulted in a premature stop codon of 24 amino acids downstream. It is expected to result in an absent or disrupted protein product. | Protein absence | 33,39 |
| p.(Pro393Ser) | rs61761869 | MWürzburg | c.1177C>T | Exon 5 | Missense mutation | Protein deficiency | 34 |
| p.(Pro393Leu) | rs199422209 | MHeerlen** (M1 Ala)MHeerlen** (M1 Val) | c.1178C>T | Exon 5 | Missense mutation | Protein deficiency | 35 |
| p.Met1? | rs1057516555 | c.1A>GQ0Attikon | c.1A>G | Exon 4 | Missense mutation | unknown | it will be fully characterized in a next paper. |
MHeerlen (M1Ala/M1Val) =p.(Pro393Leu), Q0Mattawa (M1 Val)/Q0Ourem (M3) = p.(Leu377Phefs*24),MMalton(M2)/MPalermo(M1Val)/MNichinan(V)/Q0LaPalma(S)= p.(Phe76del), PLowell (M1Val) ; PDuarte (M4), YBarcelona (p.Pro39His)= p.(Asp280Val).8
Gene-sequencing in 46.7% of patients detected Q0GraniteFalls, Q0Saint-Etienne, Q0Amersfoort (M1Ala), MWürzburg, NHartfordcity and one novel-variant (c.1A>G) named Q0Attikon. This mutation has never been described before; it relates to the codon of initiation of translation of the gene completely inhibiting AAT production. The variant was characterized as a null variant. A new name is planned in a separate paper (Table 2). Investigation of the study population by genotyping and next-generation sequencing approaches revealed that 24 out of 38 homozygous or compound heterozygous adult patients (63.2%) presented a multiplicity of rare variants and a diversity of rare combinations. PI*Q0GraniteFallsQ0GraniteFalls was encountered in 4 patients and PI*Q0Amersfoort(M1Ala)Q0Amersfoort(M1Ala) in 2, the rest of the ultra-rare combinations being represented singularly. Exceptional combinations included PI*Q0Amersfoort(M1Ala)p.(Leu377Phefs*24), (a unique one), p.(Phe76del p.(Leu377Phefs*24), and p.(Leu65Pro)p.(Pro393Leu).Genetic analysis of heterozygous patients showed 4 PI*MQ0Amersfoort(M1Ala), 1 PI*MMProcida, 1 PI*Mp.(Asp280Val) and 1 PI*MOFeyzin. Five patients were female and three ever-smokers. Age at diagnosis had a range (minimum-maximum) of 42 (34–76) years, AAT levels of 0.16 (0.62–0.78) g/L and FEV1% predicted of 64 (40–104)% (Table 3). Consanguinity was reported only once in an index-case PI*Q0GranitefallsQ0Granitefalls patient where both parents were second cousins. AAT levels were significantly different between genotypes (p = 0.002) with PI*Q0Q0 presenting the lowest levels (Fig. 3).
Clinical and molecular characteristics of adult patients heterozygous for pathogenic variants.
M=Male, F=Female, E=Emphysema, B=Bronchiectasis, BA=Bronchial asthma, FEV1= Forced expiratory volume in one second, A1AT=A1-antitrypsin, age at diagnosis (years), Ex=ex-smoker.
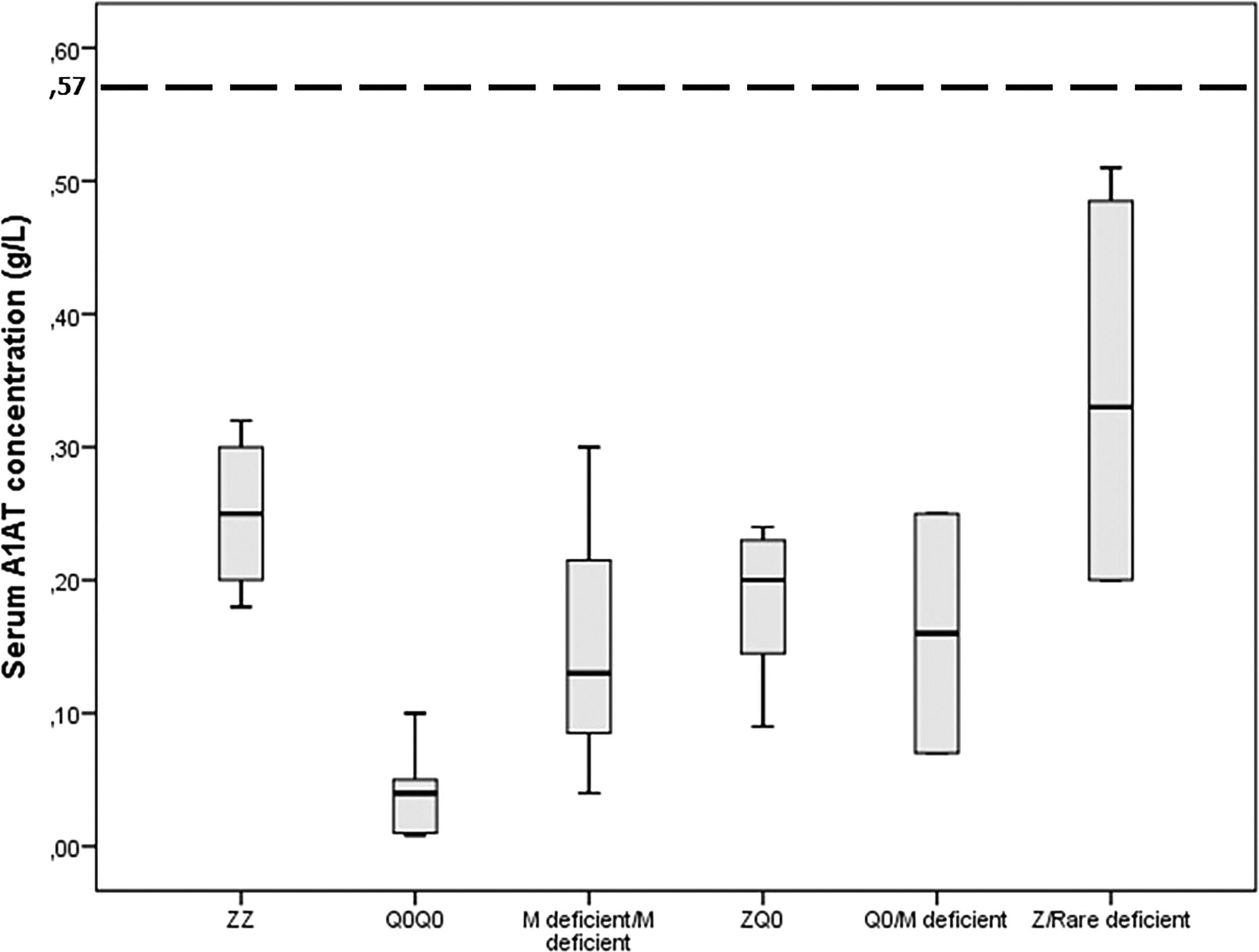
Serum A1 antitrypsin (AAT) levels in g/L for 38 homozygous or compound heterozygous for pathogenic variants Greek patients with early emphysema and AAT deficiency. Horizontal bars correspond to the median value (interquartile range) for each group based on the genetic analysis. The dashed line corresponds to the protective level of 0.57 g/L.
The present study expands knowledge about the geographical distribution of genotype variants in AATD in Europe. In Greece PI*ZZ genotype was found in only 36.8% of patients while in the rest a multiplicity of rare variants and a diversity of rare combinations, including some unique ones were observed, confirming an established South-North European geographical trend in rare variants.7,20,21 Rare variants observed in 63.2% of patients embraced the null variants PI*Q0Bellingham, PI*Q0Amersfoort, PI*Q0Granite Falls, PI*Q0Saint-Etienne, PI*Q0Mattawa and the novel PI*Q0Attikon (c.1A>G) allele; and the deficient variants PI*MHeerlen, PI*MProcida, PI*MMalton, PI*MWürzburg, and PI*NHardfordcity. All genotypes included in the study proved pathogenic since they were clinically encountered in patients with early emphysema and lower than normal AAT levels. To define genotypes 46.7% of patients required gene sequencing. PI*Q0Q0 genotype was associated with the lowest AAT serum levels.
Historically, among patients with severe AATD, the PI*ZZ genotype appeared to predominate almost exclusively (98%) in Northern Europe and in Caucasians of European descent living in the USA. A smaller although significant number of homozygous PI*ZZ patients are also found in Canada, Australia, New Zealand and some countries of South America. However, more recent studies in Europe have discovered wide geographical differences, where Northern Europe, the Iberian Peninsula and Ireland present the highest prevalence of PI*ZZ genotype, Southern and Eastern Europe the lower ones and Great Britain intermediate prevalence.20,21 In more detail and based on recent epidemiological evidence, the PI*ZZ genotype mean prevalence is 4.5 to 10 times higher in Denmark compared to countries like Serbia and North Macedonia, whereas data were completely lacking for Greece.20,21
The present study by demonstrating that in Greece only one third of patients with early emphysema and severe AATD presented the PI*ZZ genotype confirms for the first time, to the best of our knowledge in Greece, the already established North-South European geographical trend in PI*Z variant. Many theories have been developed so far to explain this observation including the Scandinavian origin of the PI*Z allele and the association of its dispersal to the Viking migration history. Updated knowledge of human genetic diversity tends to explain the observed gradients in prevalence through another perspective, the one of “South-North cline of genetic diversity”.8 Differences might result from population movements the Neolithic times from the Southern European populations historically larger and genetically more diverse to the Northern ones.22,23
The present study also expands knowledge about the geographical distribution of genotype variants in AATD in Europe by discovering in Greece a multiplicity of rare variants including six null variants (one novel one) and six rare deficiency ones in an exceptional diversity of rare combinations both homozygous and compound heterozygous rarely described before in one cohort. Interestingly the PI*S allele and the PI*SZ genotype considered as the second most prevalent ones worldwide were not represented in our study population.24 This could be partly explained by the fact that the threshold of AAT serum levels used to select affected cases could not “capture” some of those patients and that only a fraction of PI*SZ (smokers) shows respiratory symptoms and emphysema.24 In the Spanish registry of 469 patients with AATD, only 3.4% were carriers of rare variants in homozygous or compound heterozygous state.12 In the Italian registry of 422 adult subjects with severe AATD, 20.4% of patients were found with one rare deficient/null allele in combination with an PI*S or PI*Z allele or with rare deficient/null alleles, the highest so far recorded prevalence in national registries and the closest one to our findings.13 In the US AATD registry of 1129 subjects, 97% of them were PI*ZZ and only 1.8% presented rare variants such as PI*Q0GraniteFalls, PI*Q0Bellingham, PI*MMalton, PI*MHeerleen and PI*PLowell. 14 In the German registry of 548 individuals, 8.5% of patients were carriers of rare variants.15 Finally, in the French cohort of 312 patients, only 3 patients (1.1%) were found to carry a PI*Q0/Z genotype, whereas 84.6% were PI*ZZ and 5.5% PI*SZ.16The comparison of our study with others in Europe and the USA delineates the singularity of our findings showing 63.2% of AATD patients with rare, ultra-rare and almost unique variants and raises fascinating questions about migration patterns and adaptation procedures that could explain such a genetic diversity in a small but historical land of Eastern Europe, Greece. The fact that rare alleles almost segregated in a restricted geographical area, are also observed sporadically in different geographic populations both European and American provides evidence of their ancestry.25–39 In the present cohort, the identification of null, rare-deficiency as well as a novel null variant was possible thanks to DNA-sequencing technology which was the only one compared to IEF and PCR techniques that provided an accurate and unambiguous genotyping final result.
The sequencing protocol includes the seven exons of SERPINA1 gene (NM_001127701.1): 3 first exons corresponding to 5′ UTR region and the 4 coding exons. Full exons plus intronic sequences around exon (at least 30 bp intron sequences around exons) are amplified and sequenced by Next Generation Sequencing (MiSeq, Illumina).17,40 This is in accordance with the recent literature which is increasingly highlighting the importance of whole SERPINA1 gene next-generation sequencing to explain new mechanisms of AATD pathophysiology in a personalized medicine era.41,42
The clinical profile of our patients corresponded to the already described one of early emphysema even in the absence of smoke exposure.43 In addition to PI*Z, null and rare-deficiency variants are considered factors increasing the risk for the development of early lung disease.2,44,45 Pathogenicity is attributed to lower-than-normal AAT levels leading to unopposed proteolytic activity (loss of function) but also to stimulation of endoplasmic reticulum stress and inflammation pathways related to polymerization of misfolded proteins especially in some of the Mdeficient variants whose biologic and molecular behavior resembles that of PI*Z deficient ones (gain of function).46
The fact that our study also discovered rare variants in a small number of symptomatic early emphysema patients found to be heterozygous only for rare variants such as PI*Q0Amersfoort(M1Ala) in four patients and PI*MProcida, p.(Asp280Val) and PI*OFeyzin further confirms our findings showing the predominance of rare variants in the Greek population. In the literature there is very little data about heterozygotes showing that PI*MZ individuals are at increased risk to develop early disease and that PI*MQ0 subjects may present with emphysema, asthma or chronic bronchitis over 45 years of age, irrespective of their smoking habit.47–49
The major limitation of our study is that it could be subject to selection or reporting bias and thus underscore the scale of AATD in Greece. Based on international epidemiological data,1 the estimated number of cases in Greece approaches approximately 2500. Given the high under-recognition rate reported worldwide at 0.35 to 4%1 the clinically identified patients should range from 9 to 100. Considering that the populations of Spain, Italy, the USA, Germany and France are 5, 6, 33.2, 8.3 and 6.7 times greater, respectively compared to Greece, the number of 45 patients included in the study may be regarded as very comparable to the “registry-based” cohorts of the countries cited-above.12–16 Indeed in the epidemiologic study of de Serres and Blanco the authors estimate a number of 43 PI*ZZ for Greece.50 We consider that this calculation could be imprecise for the Greek population. The authors of the epidemiologic study themselves comment on the potential important limitations and bias of their calculations based on the methods of selection of the cohorts and on the fact that the analysis had not taken into consideration rare variants given the paucity of data. In fact, many AATD patients in Greece were considered by their treating physicians to be “PI*ZZ” without documentation before a specific and detailed evaluation of pathogenic variants was performed with the contribution of the expert laboratory of the University of Marburg in collaboration with another 3 expert centers in Europe for the Greek patients in the present study.
ConclusionGenotyping AATD in Greece, a multiplicity of rare variants and a diversity of rare combinations, including unique ones were observed in two thirds of patients, expanding knowledge regarding European geographical trend in rare variants. Gene sequencing was necessary for genetic diagnosis. In the future the detection of rare genotypes may help to personalize preventive and therapeutic measures.
Financial supportNone.
CRediT authorship contribution statementS.A. Papiris: Conceptualization, Visualization, Funding acquisition, Formal analysis, Methodology, Writing – review & editing, Supervision. M. Veith: Formal analysis, Methodology, Writing – original draft. A.I. Papaioannou: Formal analysis, Methodology, Writing – original draft. V. Apollonatou: Funding acquisition, Methodology, Writing – review & editing. I. Ferrarotti: Methodology, Writing – review & editing. S. Ottaviani: Methodology, Writing – review & editing. A. Tzouvelekis: Funding acquisition, Methodology, Writing – review & editing. V. Tzilas: Funding acquisition, Methodology, Writing – review & editing. N. Rovina: Funding acquisition, Methodology, Writing – review & editing. G. Stratakos: Funding acquisition, Methodology, Writing – review & editing. I. Gerogianni: Funding acquisition, Methodology, Writing – review & editing. Z. Daniil: Funding acquisition, Methodology, Writing – review & editing. L. Kolilekas: Funding acquisition, Methodology, Writing – review & editing. K. Dimakou: Funding acquisition, Methodology, Writing – review & editing. G. Pitsidianakis: Funding acquisition, Methodology, Writing – review & editing. N. Tzanakis: Funding acquisition, Methodology, Writing – review & editing. S. Tryfon: Funding acquisition, Methodology, Writing – review & editing. F. Fragopoulos: Funding acquisition, Methodology, Writing – review & editing. E.M. Antonogiannaki: Funding acquisition, Methodology, Writing – review & editing. A. Lazaratou: Funding acquisition, Methodology, Writing – review & editing. E. Fouka: Funding acquisition, Methodology, Writing – review & editing. D. Papakosta: Funding acquisition, Methodology, Writing – review & editing. P. Emmanouil: Funding acquisition, Methodology, Writing – review & editing. N. Anagnostopoulos: Funding acquisition, Methodology, Writing – review & editing. T. Karampitsakos: Funding acquisition, Methodology, Writing – review & editing. K. Vlami: Funding acquisition, Methodology, Writing – review & editing. M. Kallieri: Funding acquisition, Methodology, Writing – review & editing. P. Lyberopoulos: Funding acquisition, Methodology, Writing – review & editing. S. Loukides: Funding acquisition, Methodology, Writing – review & editing. D. Bouros: Funding acquisition, Methodology, Writing – review & editing. A. Bush: Methodology, Writing – review & editing. M. Balduyck: Formal analysis, Methodology, Writing – review & editing. C. Lombard: Formal analysis, Methodology, Writing – review & editing. V. Cottin: Formal analysis, Methodology, Writing – review & editing. J.F. Mornex: Formal analysis, Methodology, Writing – review & editing. C.F. Vogelmeier: Formal analysis, Methodology, Writing – review & editing. T. Greulich: Formal analysis, Methodology, Writing – review & editing. E.D. Manali: Conceptualization, Visualization, Funding acquisition, Formal analysis, Methodology, Writing – review & editing, Conceptualization, Visualization, Funding acquisition, Formal analysis, Supervision, Writing – review & editing.
The authors would like to acknowledge the contribution of the following collaborators in patient care: Aimilia Tsaroucha (2nd Pulmonary Department, Athens Chest Hospital "Sotiria", Athens Greece), Ilias C Papanikolaou (Department of Pulmonary Medicine, Corfu General Hospital), Michael Kitrou (Patras, Greece), Aggeliki Haritou (Ioannina, Greece), Zafeirios Sardelis (7th Pulmonary Department, Athens Chest Hospital "Sotiria", Athens Greece), Nikolaos Koulouris, Eleni Stagaki (1st Respiratory Medicine Department of the National, Kapodistrian University of Athens, Athens, Greece), Gerasimos Apollonatos (Peristeri, Athens, Greece), Maria Tsiamita (Department of Respiratory Medicine, General Hospital of Patras, University of Patras), Panagiota Loukopoulou (Aigio, Greece), David Tsoukas (Florina, Greece), Stamatia Markou (Xanthi, Greece), Panagiotis Mavroudis (Hygeia Hospital, Athens), Ioanna Korbila, Galateia Verykokou, Maria Maniati, Konstantinos Kagouridis (2nd Pulmonary Medicine Department, General University Hospital "Attikon", Medical School, National and Kapodistrian University of Athens), Konstantinos Douros, Smaragdi Fessatou, Ino Kanavaki, Vassiliki Papaevangelou (Third Department of Pediatrics "Attikon" University Hospital, National and Kapodistrian University of Athens School of Medicine Athens), Iraklis Tsangaris (Second Department of Critical Care Medicine, General University Hospital "Attikon", Medical School, National and Kapodistrian University of Athens) (all in Greece) and Julie Traclet (Service de pneumologie, centre national coordinateur de référence des maladies pulmonaires rares, Hôpital Louis Pradel, Hospices Civils de Lyon, Université de Lyon, Université Claude Bernard Lyon 1, UMR754 INRA, IVPC, Lyon, France)


