

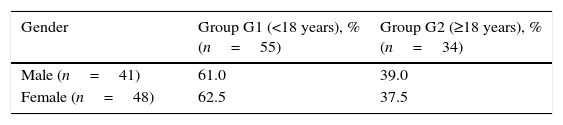
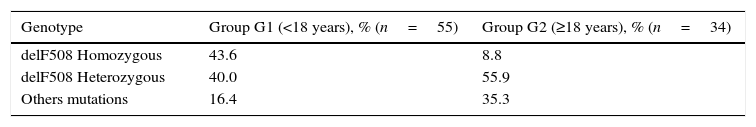

Cystic fibrosis (CF) is the most common autosomal recessive disease in Caucasians. Although most cases are diagnosed in childhood, diagnosis in adults is apparently increasing.
ObjectiveEvaluate the adult population with CF, comparing patients who were diagnosed before and after 18 years of age.
MethodsRetrospective analysis of patients followed in three main medical centres in Portugal in 2012. Comparison of two groups: G1 – patients diagnosed at <18 years and G2 – patients diagnosed at ≥18 years.
Results89 adults were identified: 61.8% in G1, 38.2% in G2. Gender distribution was similar in both groups. Average age in G2 was higher (38.3±8.4 vs. 26.8±6.1 years, p<0.001). Respiratory symptoms most frequently led to CF diagnosis in all patients, mainly in adulthood. There was a greater percentage of patients homozygous for the mutation delF508 in G1 (43.6 vs. 8.8%, p=0.02). Respiratory and pancreatic function, and body mass index (BMI) showed a higher severity in G1 (G1 vs. G2: FEV1: 54.6±27.3 vs. 29.9±64.6%, p=0.177; pancreatic insufficiency 72.7 vs. 26.5%, p<0.001; BMI 20.2±3.4 vs. 22.2±4.8, p=0.018). Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus were the most frequently isolated microorganisms. Lung transplantation rate was higher in G2 (20.6 vs. 10.9%, p=0.231) while mortality rate was higher in G1 (0 vs. 3.6%, p=0.261). Hospital admission rate was higher in G1 as well as mortality rate.
ConclusionThe results suggest that patients with CF diagnosed in childhood have characteristics that distinguish them from those diagnosed in adulthood, and these differences may have implications for diagnosis, prognosis and life expectancy.
Cystic fibrosis (CF) is the most common autosomal recessive disease affecting 1 in 2500 newborns among Caucasians.1
Its incidence is well documented in Europe where 1 in 2000–3000 newborns are affected.2 In Portugal, the incidence is apparently lower it is estimated at 1:6000 live births.3 Currently, however, there are only about 300 CF patients (approximately one-third of whom are adults) in follow-up in specialized centres, fewer than expected considering the incidence value above.
The gene responsible for CF encodes a protein of 1400 amino acids, which functions as a chloride-transport-channel located in the apical epithelial cells, thereby regulating the movement of solute and H2O.4–6 It was identified in 19897–9 and since then, more than 2000 disease-associated mutations have been described.10
The most common mutation, which is termed delF508 as a phenylalanine-residue at position 508 is absent, is present in approximately 70% of defective cystic-fibrosis-transmembrane-conductance-regulator (CFTR) alleles.10
CF is a highly variable disease and patients are diagnosed with different presentations. Symptoms appear in the first year of life in the great majority of cases but may appear later, even in adulthood with considerable variation in severity and rate of progression.4
The gold-standard laboratory method for diagnosing CF is the sweat test but there are other methods such as genetic analysis or the determination of nasal transepithelial potential difference.11,12 Since 2013 a pilot screening programme for CF, applied to all newborns, has been running in Portugal.
Patients are diagnosed when at least one of these tests positive, identification is associated with one or more phenotypic characteristics, a positive neonatal screening or family history.12
Although CF is usually discovered early in life, in recent decades it is no longer an exclusively paediatric disease. The number of adults with CF continues to increase, while the number of children has remained relatively stable over the past decade. In 2013, adults comprised 49.7% of the CF population, compared with 29.2% in 1986.13 This increase may be due to increased survival, secondary to effective treatment, more diagnostic capabilities with the inclusion of milder forms of the disease, greater availability and improved genetic testing, using more specific and sensitive diagnostic criteria, as well as increased sensitivity to the possibility of medical diagnosis.14,15
The first adult CF diagnosis was made in 1946.16 Since that time, a number of case studies of adults receiving a diagnosis have been reported, and the incidence of delayed diagnosis is expected to increase.
There are a number of reasons for diagnosis as adults. Firstly, mild expression of the disease, absent symptoms or unusual presentations may delay a patient seeking medical attention.17–19 Secondly, the common perception of CF as a disease exclusive to childhood leads to it not being recognized in adults, especially when the presenting symptoms are atypical.20,21
We have found that patients presenting in adulthood appear to be different to patients presenting in childhood. Research has established that, as a group, those diagnosed as adults have variable and atypical presentations, and often have milder disease,22,23 better, long-term prognosis,23,24 better lung function, higher rates of pancreatic sufficiency, fewer complications, and longer life expectancy than adults diagnosed in childhood.24–26
There is a growing need to identify differences between the groups but there are no published data in Portugal.
The objectives of this study were: (1) to characterize the adult CF Portuguese population, comparing patients who were diagnosed before and after 18 years of age; (2) to determine differences between the two groups regarding demographic characteristics and variables related to CF; and (3) to investigate potential diagnostic and therapeutic implications of these differences.
MethodsA retrospective analysis of clinical records of patients followed in the three main specialized adult centres in Portugal (Porto, Coimbra and Lisbon) between January 01 and December 31, 2012.
All patients were diagnosed with CF according to the diagnostic criteria in their consensus statement.
These patients were assigned to one of two groups: those diagnosed at <18 years old (Group1 – G1) and those diagnosed at ≥18 years old (Group2 – G2).
Several variables were evaluated (demographic, diagnostic, genetic, mortality, health status, and other related variables) after which the two groups were compared.
The patients were analysed into CFTR mutation classes, and according to these, were subdivided into severe (classes I, II or III) or mild (patients with at least one allele from class IV to VI).
Statistical analysis was performed using SPSS Statistics 19.0.
ResultsEighty-nine patients with CF were identified, 55 (61.8%) diagnosed at age <18 years of age (G1) and 34 (38.2%) at age ≥18 years of age (G2).
Forty-one patients (46%) were male and 48 (54%) were female. Gender distribution was similar in both groups (G1 vs. G2: women 54.5% vs. 52.9%, men 45.5% vs. 47.1%).
The average age was 31.3±9.0 years. There were statistically significant differences in relation to current age, and the average age of patients in G2 was older (38.3±8.4 vs. 26.8±6.1 years, p<0.001).
All of the included patients were Caucasian.
The median age at diagnosis was 13, and 34 (38.2%) were diagnosed in adulthood.
Over one-half of adults were women, yet men were more likely to receive a late diagnosis (Table 1).
Association between age at diagnosis and gender in patients with CF.a
| Gender | Group G1 (<18 years), % (n=55) | Group G2 (≥18 years), % (n=34) |
|---|---|---|
| Male (n=41) | 61.0 | 39.0 |
| Female (n=48) | 62.5 | 37.5 |
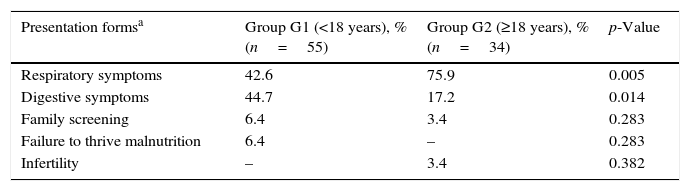
There were significant associations between age at diagnosis and the condition suggesting the diagnosis (respiratory and digestive symptoms), as noted in Table 2.
Association between age at diagnosis and condition suggesting diagnosis.
| Presentation formsa | Group G1 (<18 years), % (n=55) | Group G2 (≥18 years), % (n=34) | p-Value |
|---|---|---|---|
| Respiratory symptoms | 42.6 | 75.9 | 0.005 |
| Digestive symptoms | 44.7 | 17.2 | 0.014 |
| Family screening | 6.4 | 3.4 | 0.283 |
| Failure to thrive malnutrition | 6.4 | – | 0.283 |
| Infertility | – | 3.4 | 0.382 |
Although respiratory symptoms (recurrent/acute respiratory infections, cough, haemoptysis) were the most frequent clinical manifestations leading to the diagnosis in all patients, this is particularly evident when patients are diagnosed as adults (G2 vs. G1: 75.9% vs. 42.6%; p=0.005). Patients in G1 showed a greater variability in conditions suggesting the diagnosis, and digestive symptoms that most frequently lead to diagnosis (G1 vs. G2: 44.7% vs. 17.2%; p=0.014). In G2, 3.4% of patients were diagnosed due to fertility problems and failure to thrive was observed only in G1. Furthermore, respiratory symptoms were more frequent in female patients (70.7% vs. 37.1%, p=0.003) whereas digestive symptoms were the most frequent in males (54.3% vs. 17.1%, p<0.001).
All patients studied were genotyped. The most frequent mutation identified was delF508 (54.9%), 27 patients (30.3%) were homozygous and 41 (46.1%) heterozygous. The second most common mutation was R334W (12.7%). There was a greater percentage of patients homozygous for the mutation delF508 in G1 (43.6% vs. 8.8%, p=0.02) as illustrated in Table 3. Greater heterogeneity of mutations in the G2 has been verified; significantly higher percentages of patients in G2 were either heterozygous for delF508 or did not carry the allele (91.2% vs. 56.4%, respectively).
Association between genotype delF508 and age at diagnosis.a
| Genotype | Group G1 (<18 years), % (n=55) | Group G2 (≥18 years), % (n=34) |
|---|---|---|
| delF508 Homozygous | 43.6 | 8.8 |
| delF508 Heterozygous | 40.0 | 55.9 |
| Others mutations | 16.4 | 35.3 |
According to CFTR mutation classes, 40 (46.5%) had CFTR mutation classes I, II or III (severe) and 46 patients (53.5%) had at least one allele from classes IV–VI (mild).
Thirty-six patients (66.7%) showed severe mutation classes in G1, compared with 4 patients (11.8%) in G2 (p<0.001).
Considering pancreatic function, 39 patients (97.5%) with severe CFTR mutation classes presented pancreatic insufficiency vs. 9 patients (19.6%) with mild CFTR mutation classes (p<0.001).
The average value of FEV1 was 58.8±28.9%, 23 patients (25.8%) with FEV1>80% and 26 (29.2%) <40%, 3 of which <20%. The assessment of respiratory function showed a higher severity in G1, with patients in G2 attaining slightly higher mean percentage predicted of FVC and FEV1 scores; there was not a significant difference in FVC and FEV1 values between groups (Table 4).
Association between lung function and microbiological result with age at diagnosis.
| Lung function | Group G1 (<18 years)a | n | Group G2 (≥18 years)a | n | p-Value |
|---|---|---|---|---|---|
| FEV1% predicted | 54.6 (27.3) | 54 | 65.6 (30.6) | 34 | 0.148 |
| FVC % predicted | 72.1 (31.3) | 27 | 85.6 (29.2) | 20 | 0.197 |
| Microbiological results | Group G1 (<18 years)b | n | Group G2 (≥18 years)b | n | p-Value |
|---|---|---|---|---|---|
| Pseudomonas aeruginosa | 60.0 | 33 | 47.1 | 16 | 0.33 |
| Burkholderia cepacia | 12.7 | 7 | 0 | 0.041 | |
| Methicillin-sensitive Staphylococcus aureus | 58.2 | 32 | 52.9 | 18 | 0.792 |
| Methicillin-resistant Staphylococcus aureus | 20.0 | 11 | 14.7 | 5 | 0.728 |
| Achromobacter baumannii | 7.4 | 2 | 5.0 | 1 | 1.0 |
| Aspergillus | 22.2 | 6 | 5.0 | 10 | 0.094 |
| Nontuberculosis mycobacterium | 5.5 | 3 | 5.9 | 2 | 1.0 |
Thus, despite being considerably older (about 12 years difference), the G2 patients presented results of lung function tests which were similar to the G1.
Forty-nine patients (56%) had exocrine pancreatic insufficiency and 8 of these also had CF-related diabetes and were on insulin therapy. A higher rate of pancreatic insufficiency was found in G1 and this difference was significant (74.1% vs. 27.3%, p<0.001). Pancreatic function is closely related to the genotype. All patients with delF508 homozygous had pancreatic dysfunction.
The average value of body-mass-index (BMI) was 20.9±4.1kg/m2. Forty-one patients (46.1%) had BMI<20 and 14 (15.7%) were obese (BMI>25).
Assessment of BMI showed a higher level of this variable in G1 and this difference was statistically significant (20.1±3.4 vs. 22.2±4.8, p=0.018).
In culture result analysis, lung transplant patients (14) were excluded and therefore only 75 patients were considered. The most common microbiological isolates were Pseudomonas aeruginosa (Pa) in 49 patients (55.1%) and methicillin-sensitive-Staphylococcus-aureus (MSSA) in 50 (56.2%). Most of the patients had fewer than three microbiological isolates and more than two germs isolated. Regarding differences between microbiological isolation and age at diagnosis (Table 4), Pa and MSSA were the most frequently isolated microorganisms in both groups (G1 vs. G2: Pa: 60.0% vs. 47.1%, p=0.33; MSSA: 58.2% vs. 52.9%, p=0.792). Burkholderia cepacia was isolated only in G1 (12.7% vs. 0%, p=0.041).
Lung transplantation rate was higher in G2 (G2 vs. G1: 20.6% vs. 12.7%, p=0.490).
Fourteen-patients were hospitalized for complications. Hospital admissions rate was higher in G1 (G1 vs. G2: 44.0% vs. 17.6%, p=0.148). Two-patients (2.2%) died due to lung transplant rejection and sepsis of pulmonary origin. Mortality rate was higher in G1 (G2 vs. G1: 0% vs. 3.6%, p=0.522).
DiscussionIn consideration of the fact that diagnosis of CF in adulthood is possible and appears to be increasing,26 this study explored issues dealing with late diagnosis, with the objective of identifying any possible differences in clinical presentation of CF between patients diagnosed before and after 18 years of age.
In this study adults who received late diagnosis of CF have unique characteristics that suggest particular needs in terms of treatment and support; they experienced fewer complications and fewer hospital admissions than those who received diagnosis under 18 years of age, in whom respiratory (p=0.148) and pancreatic (p<0.001) involvement was less severe and had a significantly better nutritional status (p=0.018).
Significant association between age at diagnosis and the condition suggesting the diagnosis was found, in spite of the fact that respiratory symptoms were the most frequent manifestations leading to the diagnosis in all patients; this is particularly evident when the diagnosis is made in adulthood.
The study also found a greater percentage of homozygous delF508 among patients diagnosed earlier in life than in those diagnosed in adulthood, and this difference was statistically significant. It is recognized that homozygosity for delF508 mutation is associated with a high systemic repercussion of CF, a high level of pancreatic insufficiency and a low BMI. On the other hand, the greater heterogeneity in mutations of patients with a late diagnosis found in this study could be responsible for the greater variance in conditions leading to diagnosis and the overall better indexes of health in this group; it is a fact that patients diagnosed in adulthood had fewer hospitalizations and experience fewer complications than those diagnosed early in life and most of them were pancreatic sufficient, which is closely related to the type of CFTR mutation associated.27,28
In the study, as is common in the literature, most of those diagnosed early in life present with a combination of gastrointestinal and pulmonary symptoms, while in the late diagnosis group of patients most were diagnosed due to recurrent pulmonary infections and gastrointestinal symptoms were less frequent.25
The acquisition of infectious agents is an important environmental modifier which determines course and prognosis of most patients with CF. Furthermore, patients who are infected experience a more rapid decline of lung function than their non- infected peers.29 Although no significant difference between age at diagnosis and culture results was found, and there was not a significant difference in FVC and FEV1 values between groups, there was a higher percentage of microbiological isolations in G1 patients, and the assessment of respiratory function showed a greater severity in these patients.
No significant differences were found between groups in terms of transplantation and mortality rates.
The above results provide empirical support that supports the argument that patients with a late diagnosis of CF are different from those who receive early diagnosis.
ConclusionsIn summary, this study is the first description of the Portuguese adult CF population and the first to examine two distinct populations on the basis of age at diagnosis, which is particularly important, since it gives us a better understanding of real life.
A significant percentage of our patients were diagnosed in adulthood, which highlights the need for diagnostic suspicion in a patient with chronic lung disease and atypical symptoms. In the study performed, a 10 years longer lifetime was detected in patients diagnosed in adulthood. This difference may be associated with higher frequency of homozygosity for the mutation delF508 found in patients diagnosed early in life which is usually associated to a higher systemic repercussion of CF and a higher level of pancreatic insufficiency cases and lower BMI.
The results suggest that patients diagnosed in the paediatric age group have characteristics that distinguish them from those diagnosed in adulthood, which may have implications in terms of diagnosis, prognosis and life expectancy.
Ethical responsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors of this study would like to thank all of their colleagues at the Centro Hospitalar de São João, Centro Hospitalar de Lisboa Norte (Hospital de Santa Maria) e dos Hospitais da Universidade de Coimbra (Centro Hospitalar e Universitário de Coimbra) who contributed in this study.


