


Idiopathic Pulmonary Fibrosis (IPF) is the most common disease in the subgroup of idiopathic interstitial pneumonias. It is inevitably associated to a bad prognosis, although assuming a highly variable clinical course.
MethodsPatients with IPF, observed at Interstitial Lung Diseases outpatient clinic of Centro Hospitalar de São João – Porto, Portugal, were identified and clinical, functional, radiological and bronchoalveolar lavage (BAL) parameters were reviewed. Their clinical course and survival were analyzed in order to identify prognostic factors.
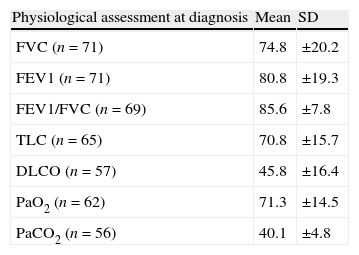
ResultsEighty-one patients were included, with a mean age at diagnosis of 63.8 years old. At diagnosis, the main functional abnormalities were restrictive physiology, reduced lung diffusion and exercise capacity impairment. Clinical course was mainly slowly progressive (72.3%). Ten patients (13.2%) had a rapid progression and 11 (14.5%) patients had an acute exacerbation during the course of the disease. IPF's rapid progression was associated to a higher functional impairment at diagnosis, namely in what is related with Functional Vital Capacity (FVC) and Total Lung Capacity (TLC). Median survival was 36 months. A significant difference in survival was observed among different types of clinical course – 41 months for slow progressors and 9 months for rapid progressors. Lower levels of FVC, TLC, six-minute walk test (6MWT) distance and rest PaO2, and higher BAL neutrophil count were associated with poorer survival in univariate analysis.
ConclusionThe analysis of this group of IPF patients confirms two clearly different phenotypes, slow and rapid progressors. Those phenotypes seem to have different presentations and a remarkably different natural history. These results could mean different physiopathologic pathways, which could implicate different therapeutic approaches.
A Fibrose Pulmonar Idiopática (FPI) é a patologia mais comum no subgrupo das Pneumonias Intersticiais Idiopáticas. Apesar de uma grande variabilidade no tipo de evolução clínica, está inexoravelmente associada a um mau prognóstico.
Material e métodosForam identificados doentes com FPI, observados na consulta de Doenças Pulmonares Difusas do Centro Hospitalar de São João, Porto, e revistos os seus parâmetros clínicos, funcionais, radiológicos e do lavado broncoalveolar (LBA). A evolução clínica e sobrevivência foram avaliadas, tendo sido igualmente identificados factores prognósticos.
ResultadosForam incluídos 81 doentes com uma média de idade de 63.8 anos. Na altura do diagnóstico, as principais alterações funcionais identificadas foram restrição, redução da difusão pulmonar e da capacidade de exercício. A maioria dos doentes (72.3%) apresentou uma evolução clínica lentamente progressiva. Em 10 doentes (13.2%), foi observada uma evolução rapidamente progressiva e 11 (14.5%) apresentaram exacerbação aguda. Verificou-se uma associação entre a evolução rapidamente progressiva e uma maior gravidade funcional ao diagnóstico, nomeadamente na Capacidade Vital Forçada (CVF) e Capacidade Pulmonar Total (CPT). A sobrevivência mediana foi de 36 meses. Verificou-se uma diferença estatisticamente significativa na sobrevivência entre os diferentes grupos de evolução clínica - 41 meses para os doentes com evolução lentamente progressiva e 9 meses para os doentes com evolução rapidamente progressiva. Valores inferiores de CVF, CPT, distância na Prova da marcha dos 6 minutos e PaO2 em repouso, bem como o maior grau de neutrofilia no LBA estiveram associados a uma sobrevivência inferior em análise univariada.
ConclusãoA análise deste grupo de doentes com FPI confirma a existência de 2 fenótipos claramente distintos, o de evolução lenta e o de evolução rapidamente progressiva. Estes fenótipos têm uma diferente apresentação clínica e uma história natural da doença claramente distinta, sugerindo a presença de diferentes mecanismos fisiopatológicos, os quais poderão implicar diferentes abordagens terapêuticas.
Idiopathic Pulmonary Fibrosis (IPF) is the most common disease in the subgroup of Idiopathic Interstitial Pneumonias.1,2 It is a disease of the elderly, with presentation occurring usually in the sixth and seventh decades, more frequently in men.3–5 Both etiology and key mechanisms of pathogenesis remain to be understood.3
IPF's typical histological pattern is Usual Interstitial Pneumonia (UIP).6 However, in the absence of biopsy, IPF can be diagnosed based on ATS/ERS criteria – a group of clinical, radiological and physiological parameters, internationally accepted and validated, in which typical signs at Chest High-Resolution Computerized Tomography (HRCT) have a prominent role.3,7
The clinical history of the disease is quite variable; there is usually a slow physiological deterioration, but in some patients there is a faster decline in lung function and death occurs within 6–12 months after diagnosis.8–10 Others experience an acute exacerbation during the course of the disease with a sudden worsening of respiratory symptoms, hypoxemia and the appearance of new radiological infiltrates without an identifiable cause.11,12
Despite different types of clinical course, IPF is inevitably associated with a poor prognosis, with a median survival of 2–3 years.8,13 No proven effective pharmacological therapy has yet been found.3 Some prognostic factors have already been described, which may have implications for potential therapy, particularly when to refer for lung transplant, which up until now has been the only therapy with demonstrated survival benefit.14,15 Recently, Pirfenidone, an oral antifibrotic and anti-inflammatory drug, has been approved by EMEA (European Medicines Agency) for the treatment of IPF.
Our aim was to describe the clinical presentation and the course of IPF patients who have been evaluated in recent years in our Interstitial Lung Diseases (ILD) outpatient clinic and analyze factors associated with survival and the clinical course.
MethodsPatients diagnosed with IPF, who attended an ILD outpatient clinic in Hospital de São João, a tertiary reference center in Oporto, Portugal, were identified. IPF diagnosis was obtained through an UIP compatible histology by surgical lung biopsy in 25 patients (30.9%). In 56 patients (69.1%), IPF diagnosis was based on ATS/ERS criteria (according to 2002 consensus).2 Whenever atypical signs were found on HRCT or other atypical characteristics that could raise diagnostic issues, patients were sent for surgical lung biopsy. Patients whose UIP pattern could be explained by other diseases (connective tissue diseases, hypersensitivity pneumonitis, drug lung toxicity and other ILDs whose final stage is fibrosis), were excluded.
Medical records were retrospectively analyzed. Patients were characterized clinically, physiologically and by radiological findings, at the time of diagnosis.
Rest and exercise physiological assessments (spirometry, lung diffusing capacity for carbon monoxide – DLCO, Total Lung Capacity – TLC, six-minute walk test – 6MWT and cardiopulmonary exercise test) were measured according to ERS and ATS recommendations16–20 and results were expressed as percentages of the normal predicted values. Physiological parameters obtained after any medication that could probably modify the course of the disease were excluded.
Some patients were not able to perform the exercise capacity evaluation due to a high degree of disability at diagnosis.
All patients performed HRCT scan at diagnosis. As this was a retrospective study, some of the HRCT images were not available for analysis, although all the reports were accessible. For imaging characterization, chest HRCT images were analyzed by two radiologists and the extent of the disease was calculated as a fibrotic score,21 the same used to validate the Composite Physiologic Index (CPI).
Bronchoalveolar lavage (BAL) was performed on an outpatient basis. At the time of BAL, no symptoms or signs suggestive of respiratory infection or exacerbation of the disease were observed. The performance and processing of BAL was carried out according to guidelines of the ERS Task Group on BAL.22 Total and differential cell count was classified according to the values proposed by ERS Task Group on BAL (lymphocytosis>15%, neutrophilia>3%, eosinophilia>0.5%).22
Open lung biopsy was performed under general anesthesia and the most common approach used was a limited anterolateral thoracotomy via a 4±10cm submammary incision, which allowed access to segments of different lobes in order to obtain multiple biopsy specimens. Open lung biopsy was always performed through thoracotomy. The site of the biopsy was decided on the basis of the HRCT scan. Biopsy samples were observed and evaluated independently by two pathologists.
To evaluate the occurrence of pulmonary hypertension, all patients performed echocardiography. Only the patients referred for lung transplantation underwent right heart catheterization.
Clinical course was classified as slow progression, rapid progression and acute exacerbation (AE-IPF). Rapid progression was differentiated from slow progression by a shorter duration of symptoms at diagnosis (<6 months) and a more rapid clinical deterioration.10 AE-IPF was defined as a sudden worsening of respiratory symptoms with hypoxemia and appearance of new radiological infiltrates without an identifiable cause.11
Patients included had IPF diagnoses established between 2000 and 2010. They were followed during the course of the disease, until death or lung transplant.
Statistical analysisKolmogorov–Smirnov test was used to check for normality of the distributions in all continuous variables. Medians for covariates and percentages of factors were computed and baseline differences in patients with slow versus rapid progression were assessed through U Mann–Whitney, Chi square or Fischer exact test, as appropriate.
Median survival was estimated using Kaplan–Meier survival curves. Patients were censored at the time of death or lung transplant. Follow-up was stopped after 48 months, to increase statistical power of the study. To identify the strength of prognostic factors, Hazard Ratios (and their respective 95% confidence intervals) were computed using univariate Cox regression models.
All analyses were performed using SPSS® software v. 18. A p-value<0.05 was considered significant.
ResultsPatients’ characterization at diagnosisEighty-one patients were included, 56 (69.1%) men and 25 (30.9%) women. Mean age at diagnosis was 63.8±10.2 years old. Two patients (2.5%) had familial IPF. A smoking history was found in 51.9% (27.5% current and 72.5% former smokers) accounting for a mean of 37.5 pack-years. The main symptom at presentation was dyspnea (96.3%), followed by cough (77.9%). Median time between beginning of symptoms and time of diagnosis was 12 months.
Restrictive physiology and reduced lung diffusion capacity were the main functional abnormalities (Table 1) as well as exercise capacity impairment. 6MWT was taken into consideration for 40 patients, with a mean (Standard deviation – SD) distance of 369.6 (149.4) meters and a mean (SD) lowest oxygen saturation of 81.5 (8.1) %. Cardiopulmonary Exercise Test was looked at for only 15 patients with a mean (SD) peak oxygen consumption of 66.6 (18.4)%.
HRCT scan fibrotic score was measured in 42 patients. Mean (SD) fibrotic score was 10.8 (2.3).
The prevalence of emphysema in this IPF cohort was 26.4% (14/53 HRCT available for reevaluation). Patients with emphysema were younger – 61.8 vs. 69.9 years (p=0.042), had a higher smoking load – 28.3 vs. 10.7 pack-years (p=0.035), lower FEV1/FVC – 79 vs. 86.9% (p=0.001) and more severely compromised diffusion capacity (DLCO) – 36.8 vs. 49.9% (p=0.016).
BAL was performed in 56 patients at the time of diagnosis, with neutrophilia (mean=10.8, SD=2.3) in 52 (92.9%) patients, eosinophilia (mean=5.1, SD=5.0) in 48 (85.7%) patients and mild lymphocytosis being detected in 14 (25%) patients (mean=12, SD=7.8).
Clinical courseRegarding therapeutic approach, ATS/ERS statement was followed and, if there were no contraindications, low doses of corticosteroids (86.4%) and immunosuppressive drugs (65.4%), preferably azatioprine or alternatively, cyclophosphamide were used. After the publication of IFIGENIA trial23 in 2005, N-acetylcysteine (1800mg/day) was added to this regimen (51.9%). Up until now, Pirfenidone has not been available in Portugal. Moreover, when the included IPF patients were diagnosed, Pirfenidone was still not approved by EMEA and there was no Portuguese center participating in any clinical trial using this drug.
Since a protocol was established in 2007 with Hospital Juan Canalejo (Coruña) and Hospital Santa Marta (Lisbon), patients under 65 years have been considered to lung transplantation. As this series includes patients diagnosed since 2000, there is still only a small percentage of transplanted patients. To be precise, 9 patients (7.4%) were selected for lung transplantation and single lung transplant was performed in 6 (2 patients were excluded because of infection by Aspergillus fumigatus). Another patient is on waiting list.
Clinical course (n=76) was mainly slowly progressive (72.3%). In 10 patients (13.2%), IPF had a rapid progression and 11 (14.5%) patients had an AE-IPF during the course of their disease.
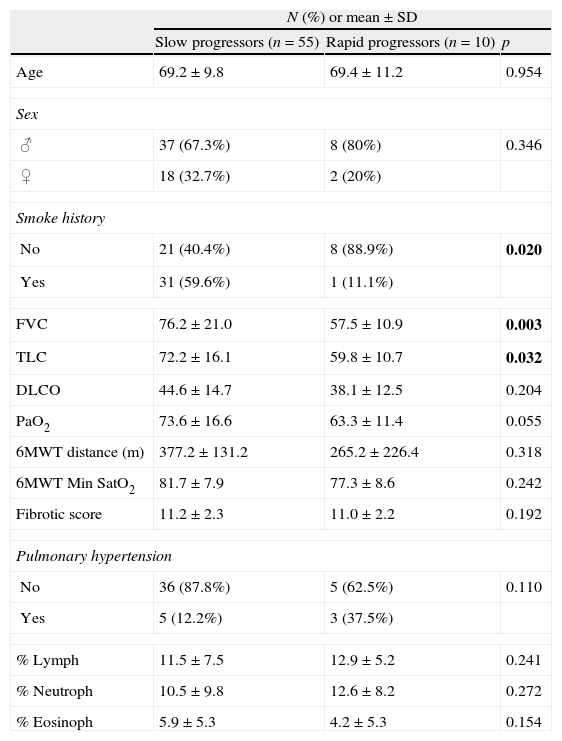
IPF's rapid progression (Table 2) was found to be associated to a higher functional impairment at diagnosis, namely with FVC (p=0.003) and TLC (p=0.032). A smoking history was associated with a slowly progressive clinical course (p=0.020).
Baseline factors and its association with clinical course (rapid versus slow progression).
| N (%) or mean±SD | |||
| Slow progressors (n=55) | Rapid progressors (n=10) | p | |
| Age | 69.2±9.8 | 69.4±11.2 | 0.954 |
| Sex | |||
| ♂ | 37 (67.3%) | 8 (80%) | 0.346 |
| ♀ | 18 (32.7%) | 2 (20%) | |
| Smoke history | |||
| No | 21 (40.4%) | 8 (88.9%) | 0.020 |
| Yes | 31 (59.6%) | 1 (11.1%) | |
| FVC | 76.2±21.0 | 57.5±10.9 | 0.003 |
| TLC | 72.2±16.1 | 59.8±10.7 | 0.032 |
| DLCO | 44.6±14.7 | 38.1±12.5 | 0.204 |
| PaO2 | 73.6±16.6 | 63.3±11.4 | 0.055 |
| 6MWT distance (m) | 377.2±131.2 | 265.2±226.4 | 0.318 |
| 6MWT Min SatO2 | 81.7±7.9 | 77.3±8.6 | 0.242 |
| Fibrotic score | 11.2±2.3 | 11.0±2.2 | 0.192 |
| Pulmonary hypertension | |||
| No | 36 (87.8%) | 5 (62.5%) | 0.110 |
| Yes | 5 (12.2%) | 3 (37.5%) | |
| % Lymph | 11.5±7.5 | 12.9±5.2 | 0.241 |
| % Neutroph | 10.5±9.8 | 12.6±8.2 | 0.272 |
| % Eosinoph | 5.9±5.3 | 4.2±5.3 | 0.154 |
% Lymph – percentage of lymphocytes at BAL differential count; % Neutroph – percentage of neutrophils at BAL differential count; % Eosinoph – percentage of eosinophils at BAL differential count. Significant results are presented in bold.
Most IPF patients (69.6%) died of causes directly related to the disease – respiratory failure, while in 13 patients (23.2%), the cause of death was due to other causes. Four patients (7.1%) died of post-lung transplant complications.
AE-IPF occurred in 11 patients. Among them, 6 (54.5%) died during the episode and 2 were submitted to lung transplantation, dying due to post-transplant complications.
SurvivalMedian survival was 36 months (Fig. 1). Twenty-five patients were still alive at the time of evaluation.
 and for slow and rapid progressors (right) – months.")
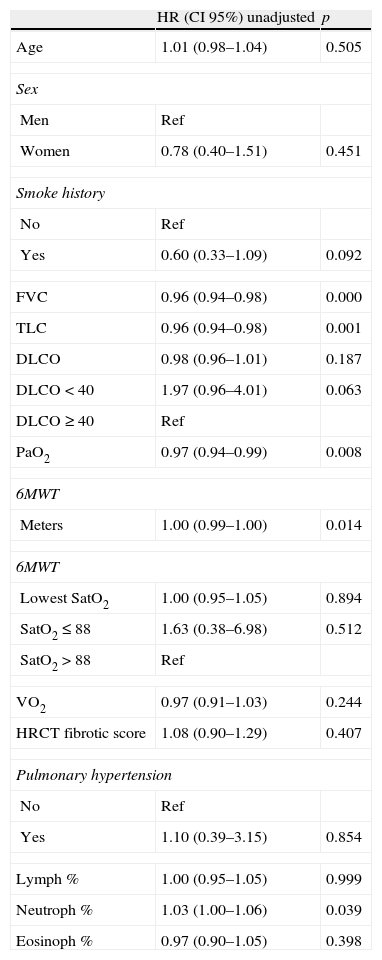
Factors found to be associated with poorer survival, in univariate analysis (Table 3), were lower levels of FVC (p=0.000), TLC (p=0.001), 6MWT distance (p=0.014) and rest PaO2 (p=0.008), as well as higher BAL neutrophil count (p=0.039).
Survival Prognostic Factors of IPF patients.
| HR (CI 95%) unadjusted | p | |
| Age | 1.01 (0.98–1.04) | 0.505 |
| Sex | ||
| Men | Ref | |
| Women | 0.78 (0.40–1.51) | 0.451 |
| Smoke history | ||
| No | Ref | |
| Yes | 0.60 (0.33–1.09) | 0.092 |
| FVC | 0.96 (0.94–0.98) | 0.000 |
| TLC | 0.96 (0.94–0.98) | 0.001 |
| DLCO | 0.98 (0.96–1.01) | 0.187 |
| DLCO<40 | 1.97 (0.96–4.01) | 0.063 |
| DLCO≥40 | Ref | |
| PaO2 | 0.97 (0.94–0.99) | 0.008 |
| 6MWT | ||
| Meters | 1.00 (0.99–1.00) | 0.014 |
| 6MWT | ||
| Lowest SatO2 | 1.00 (0.95–1.05) | 0.894 |
| SatO2≤88 | 1.63 (0.38–6.98) | 0.512 |
| SatO2>88 | Ref | |
| VO2 | 0.97 (0.91–1.03) | 0.244 |
| HRCT fibrotic score | 1.08 (0.90–1.29) | 0.407 |
| Pulmonary hypertension | ||
| No | Ref | |
| Yes | 1.10 (0.39–3.15) | 0.854 |
| Lymph % | 1.00 (0.95–1.05) | 0.999 |
| Neutroph % | 1.03 (1.00–1.06) | 0.039 |
| Eosinoph % | 0.97 (0.90–1.05) | 0.398 |
HR – Hazard ratio.
It was not possible to identify cut-points for each prognostic factor because of the lack of statistical power (small sample size).
Testing cut-points of the baseline prognostic factors described in the last consensus document3 (DLCO<40% of predicted; desaturation≤88% during 6MWT; presence of pulmonary hypertension), had no significant associations in this sample, although estimates follow the expected direction.
No significant difference in survival rates was found between IPF patients with and without emphysema.
Median survival was 41 months in slow progressors and 9 months in rapid progressors (Fig. 1).
Among slow progressors, lower levels of FVC (p=0.001), TLC (p=0.012) and 6MWT distance (p=0.026) were associated to a reduced survival.
No prognostic factors were found in rapid progressors (reduced statistical power due to a small number of patients).
DiscussionThe evaluation of this cohort of IPF patients from the north of Portugal showed the usual features that characterize this disease, in terms of clinical, functional, radiologic parameters and outcome. Although some baseline prognostic factors were associated with overall survival in this series, the main issue is related with the sort of clinical course, more exactly the distinction between slow progression and rapid progression since their outcome and survival are significantly different.
Mean age at diagnosis was consistent with literature,3,24 as well as the higher prevalence of males. Familial IPF may be undervalued. In fact, some patients report family deaths from unspecified respiratory diseases. Cigarette smoking is strongly associated with IPF, namely in those patients with heavy smoking history, more precisely more than 20 pack years.25,26 In this study, the proportion of smokers and former smokers was lower than expected, as was the case in other series.27,28 However, in the 15 patients aged under 60 years old, 80% had a history of cigarette smoking, so we can speculate that excluding ageing, smoking history could have been a significant risk factor for IPF in this sample. Twelve months was the median time between beginning of symptoms and diagnosis. Kim et al.1 reported a period of 6 months to 2 years of symptoms preceding diagnosis. In fact, not only patients tend to attribute the initial breathlessness to ageing, but also primary care physicians tend to focus the clinical investigation on cardiac diseases and other respiratory diseases such as COPD, based on their higher prevalence. However, in this cohort of patients there was an increasing number of patients referred by general practice over the last few years, which could be related to the widespread use of CT scans.
Recently, a new syndrome has been described – Combined Pulmonary Fibrosis and Emphysema (CPFE), resulting from the association of IPF and emphysema.29,30 The percentage of CPFE found in this cohort is in line with other publications.30 In this study, some previously described distinctive features were observed, namely a stronger association with smoking, lower levels of FEV1/FVC and a higher compromise on diffusion capacity (DLCO). However, some authors describe a negative impact on prognosis when IPF is associated with emphysema.30 This negative impact is strongly associated to pulmonary hypertension.30 In this study we did not find a significant difference in survival or in the occurrence of pulmonary hypertension.
Lymphocytosis was detected in 14 patients (25%). They presented a mild lymphocytosis (mean of 22.8%) and no exposure to suspected agents was detected. Also, in 50% of these patients a lung biopsy was performed to rule out other differential diagnoses. Moreover no difference on survival was found between patients with or without BAL lymphocytosis.
Clinical course cannot yet be predicted accurately. In this sample, 13.2% of patients experienced an accelerated decline, already described in literature.10 However, criteria for classification of IPF progression are not well established. The definition of rapid progressors is based on a time-period between symptoms and diagnosis of less than 6 months, associated with an accelerated decline.1,10 The difference in survival between low and rapid IPF progressors is remarkable (41 and 9 months, respectively). Although this subject is still not clearly understood, these groups must represent different phenotypes in IPF. At a univariate analysis, lower levels of FVC and TLC were associated to a rapidly progressive disease. Non-smokers were also associated to rapid progression. This was already described in other studies, also with a better survival in current smokers;31 and there is no clear explanation for this. Some data suggest that cigarette smoke inhibits lung fibroblast proliferation as well as chemotaxis, and may that way impair lung repair after lung injury.32 A recent study contradicts this fact, showing a better outcome for non-smokers, and assuming that the better outcome previously reported for smokers was compatible with a healthy smoker lead-time effect (reflecting a less severe disease at presentation).33
AE-IPF has been a main focus of interest. The results of this study are in line with publications on this subject, both in terms of incidence and mortality. In fact, incidence of AE-IPF is 10–15% of all IPF patients, according to two large randomized clinical trials 34,35 and its mortality is high, ranging between 50 and 100%.12,36–38 Excluding patients who received lung transplant, this study registered an in-hospital mortality of 66.7% in AE-IPF. There have been several attempts to identify acute exacerbations predictors, without success.38 In fact, the occurrence of AE-IPF does not appear to be related to the severity of pulmonary function impairment.38 It was the same for this study, no factor was identified which could predict its occurrence.
Median survival in this sample is in line with other studies.39,40 Predictors of mortality in IPF have been divided in baseline and dynamic predictors.41 In fact, prognosis in IPF should involve the integration of baseline and longitudinal data. This study only meant to evaluate baseline predictors. Factors associated with a poorer survival in this cohort at univariate analysis are related to higher functional decline (FVC, TLC), worse oxygenation (PaO2), reduced tolerance to exercise (distance in 6MWT) and higher neutrophil count on BAL.
When reviewing the literature, what is remarkable is the great variability in prognostic factors. There are several reasons for this, such as series with a small number of patients or the inclusion of patients with co-morbidities which act as confounding factors. Most studies have different diagnostic and therapeutic approaches relating to the patients included; although when we consider the lack of effective pharmacologic therapeutics, this latter factor is hardly likely to affect the accuracy of the results. Moreover this cohort of patients was divided according to their phenotypes, since the prognostic factors must be adapted to each of the three types of outcome as their outstanding different clinical course. The prognostic factors concerned, that is lower levels of FVC, TLC and 6MWT distance, are all significantly associated with the low progression subgroup. The lack of any association with rapid progressors may be explained by the fact that the sample size was too small to give a significant result.
Several studies have tended to ascribe prognostic values to baseline pulmonary function tests, giving the impression that a higher functional decline is significantly associated with a reduced survival. The factors concerned are usually FVC27,42–44 and TLC,42,44 with higher evidence supporting the first parameter. The association of DLCO with reduced survival rates is a consistent finding across the literature.42–45 It is a fact that lower diffusion capacity reflects a greater extent of fibrosis and increased severity of the disease. However, in this sample there was no prognostic impact. There are some studies which show baseline PaO2 at rest as a predictor of mortality,8,27,43,46 although there are also some contradictory results.44,47 Higher degrees of hypoxemia occur in more advanced stages of the disease but whether this can be considered as a prognostic factor or not, is yet to be established. There are studies which have ascribed prognostic significance in relation to survival rates to 6MWT parameters,48–51 either linked to the walking distance or to desaturation peak, but these results are underestimated due to lack of standardization and reproductability of this test.
BAL neutrophilia has been associated with severity as in other diffuse lung disease such as sarcoidosis.52 There is also some evidence that a higher level of neutrophilia may be associated with lower survival rates.53 Although neutrophilia is usually described as a baseline prognostic factor in univariate analysis, multivariate analysis does not confirm this association according to studies published in peer-review literature.54,55
Pulmonary hypertension should have a multifactorial pathogenesis for IPF. Several studies point out pulmonary hypertension to be a predictor of mortality,56,57 but usually in longitudinal studies. In our sample, pulmonary hypertension at baseline did not act as a prognostic factor.
Some coefficients21,58 have been proposed in order to combine different parameters. For instance, the Composite Physiologic Index, which includes FVC, FEV1, DLCO and fibrosis extension on HRCT scan, has been successfully tested at baseline and longitudinal studies.21,59 This could be a way to predict outcome more accurately and to provide a more precise timing of the progression of the disease.
In conclusion, IPF is inevitably associated with a poor prognosis. However, different phenotypes seem to be emerging, based on the striking differences in clinical course. A more accurate prediction of the outcome at the moment of diagnosis would improve patient management, particularly in the time to reference to lung transplant, which is the only therapy with demonstrated survival benefit.
Conflicts of interestThe authors have no conflicts of interest to declare.
Special thanks to Carla Damas, responsible for the Lung Transplant outpatient clinic of Hospital de São João, for the near cooperation on IPF patients’ referral to transplant, when indicated.
Please cite this article as: Pires FS, et al., Fibrose Pulmonar Idiopática – Apresentação clínica, evolução e factores prognósticos basais numa coorte portuguesa. Rev Port Pneumol. 2010. http://dx.doi.org/10.1016/j.rppneu.2012.05.002.


