


Interstitial lung disease (ILD) is a group of disorders characterised by chronic lung inflammation and/or fibrosis. Idiopathic pulmonary fibrosis (IPF) is one of the most common and devastating forms of ILD and is frequently diagnosed at an advanced stage, when patients have a life-expectancy of 2-3 years. There is growing evidence that genetic factors involved in IPF also contribute to a broader range of ILDs and interstitial lung abnormalities (ILA).1 ILA is defined as incidental findings of specific computed tomography features that affect more than 5% of any lung zone and are potentially compatible with ILD (i.e. ground glass or reticular abnormalities, traction bronchiectasis, honeycombing and non-emphysematous cysts).2 A growing number of studies have reported evidence of ILAs in clinically unaffected relatives of IPF patients and suggest that screening first-degree relatives is a worthwhile approach to improve detection of ILD in its early stages.3-5 Indeed, the recent Fleischner Society's position paper recommends that the identification of ILA in familial ILD should not be considered incidental and suggests use of the term “preclinical ILD”,2 and this terminology has been adopted here. We have established a clinically-annotated familial ILD genetic resource aiming to better understand disease causation. Here, we provide additional evidence in support of screening relatives of ILD patients to identify those with preclinical ILD.
We defined recruitment criteria as families with multiple first-degree relatives diagnosed with ILD or ILA, where at least one had been diagnosed with IPF (hereafter collectively termed “patients”). Families were identified via a questionnaire sent to all living Australian IPF Registry6 participants (September-November 2019) or directly recruited by respiratory physicians. Affected living individuals meeting the criteria for inclusion were recruited, in addition to at least one self-reported unaffected first-degree relative (hereafter known as “relative”). Relatives included were ≥ 50 years, except for those families with a case diagnosed before the age of 50, where relatives were included if at least as old as the youngest diagnosed case. This study reports all families in which at least one relative participated in a clinic visit. Of the families where a relative was not recruited, 38% did not have a suitably aged, living relative, whilst others were unable to participate due the impact of the global pandemic.
Informed consent and a physical examination were conducted by respiratory physicians, after which participants completed: questionnaires, including the St. George's Respiratory Questionnaire (SGRQ)7; a chest high resolution computed tomograph (HRCT) scan; full pulmonary function testing (PFT); and blood/saliva sample collection. To screen relatives for preclinical ILD, HRCT images were blinded and assessed by an ILD physician (JM). Preclinical ILD was classified using the Fleischner Society's ILA definition.2
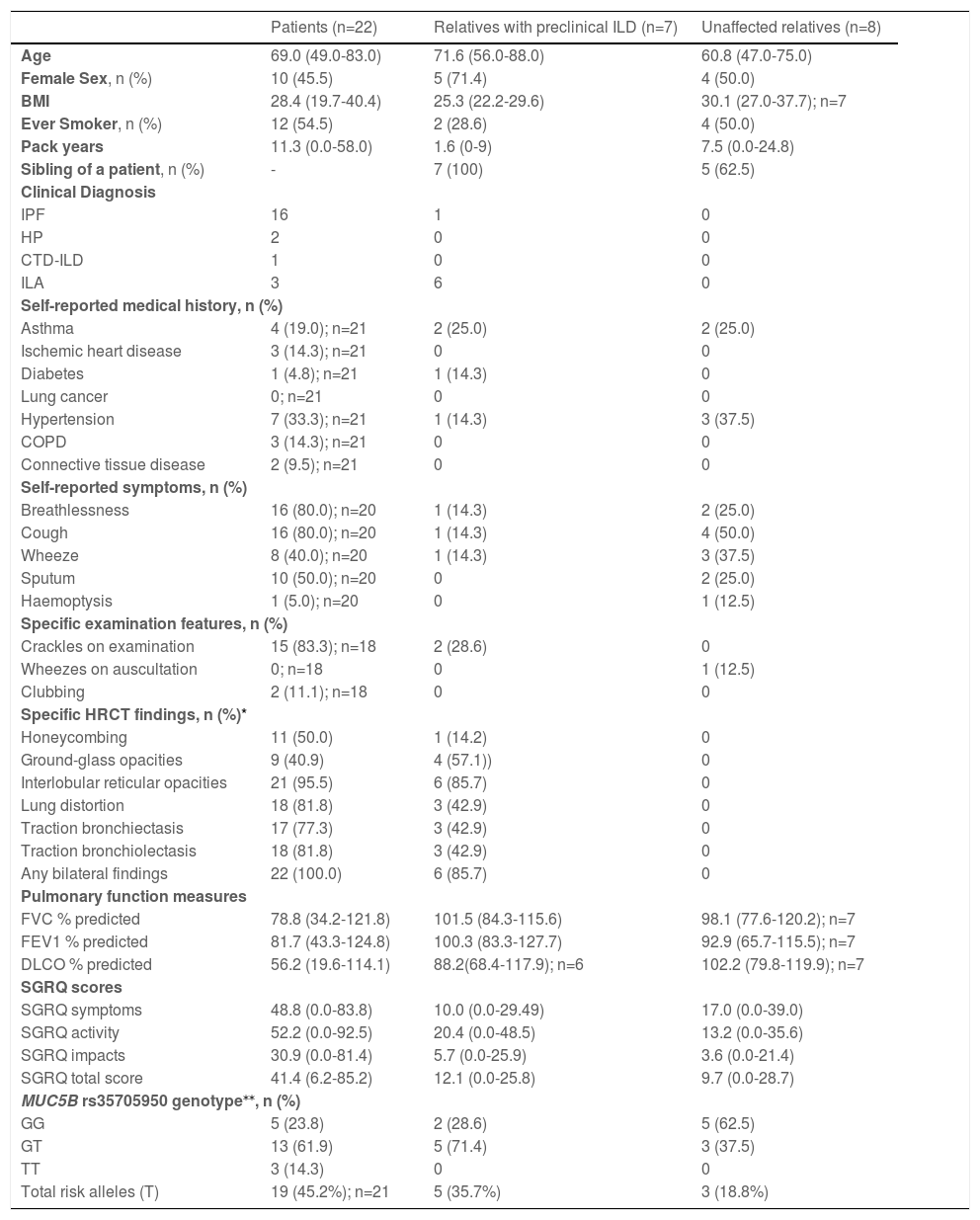
Fifteen relatives participated from 12 families (example, Fig. 1). This included ten siblings, three offspring of patients, and two individuals who had both an affected parent and sibling. Seven relatives were found to have preclinical ILD (46.7%), while eight (53.3%) had no evidence of abnormalities (Table 1). The prevalence of preclinical ILD in our Australian cohort is higher than recent studies in the American families described by Hunninghake et al.3 (26%) and Salisbury et al.5 (23%). This may be due to our strict definition of familial ILD with a confirmed family history (as opposed to self-report), although our cohort was also smaller and older (65.8 years compared to 60 years3 and 53.1 years5), which may have contributed to these differences.
; the proband is indicated by the black triangle; deceased individuals are indicated by a diagonal slash and the age range at death is indicated below followed by a ¿; grey shading indicates that the clinical diagnosis was reported by the living generation; black shading indicates participants confirmed to have IPF/ILD and participants diagnosed with preclinical ILD are indicated by half black shading; age range at examination for the unaffected relative is indicated below unshaded symbol; ‘Dx’ indicates age range at diagnosis; two living, unscreened female relatives are currently 64-69 years of age; self-reported smoking status is indicated and pack years is presented in brackets; MUC5B rs35705950 genotype is presented at the top right, G/T indicates a heterozygous carrier and G/G indicates homozygous wildtype; representative chest HRCT images are presented below study participants.")
A simplified pedigree depicting a family in which two relatives were screened: one was diagnosed with preclinical ILD while the other was unaffected at the time of examination. Males and females are depicted by squares and circles (respectively); the proband is indicated by the black triangle; deceased individuals are indicated by a diagonal slash and the age range at death is indicated below followed by a ¿; grey shading indicates that the clinical diagnosis was reported by the living generation; black shading indicates participants confirmed to have IPF/ILD and participants diagnosed with preclinical ILD are indicated by half black shading; age range at examination for the unaffected relative is indicated below unshaded symbol; ‘Dx’ indicates age range at diagnosis; two living, unscreened female relatives are currently 64-69 years of age; self-reported smoking status is indicated and pack years is presented in brackets; MUC5B rs35705950 genotype is presented at the top right, G/T indicates a heterozygous carrier and G/G indicates homozygous wildtype; representative chest HRCT images are presented below study participants.
Clinical characteristics of participants at the time of recruitment
| Patients (n=22) | Relatives with preclinical ILD (n=7) | Unaffected relatives (n=8) | |
|---|---|---|---|
| Age | 69.0 (49.0-83.0) | 71.6 (56.0-88.0) | 60.8 (47.0-75.0) |
| Female Sex, n (%) | 10 (45.5) | 5 (71.4) | 4 (50.0) |
| BMI | 28.4 (19.7-40.4) | 25.3 (22.2-29.6) | 30.1 (27.0-37.7); n=7 |
| Ever Smoker, n (%) | 12 (54.5) | 2 (28.6) | 4 (50.0) |
| Pack years | 11.3 (0.0-58.0) | 1.6 (0-9) | 7.5 (0.0-24.8) |
| Sibling of a patient, n (%) | - | 7 (100) | 5 (62.5) |
| Clinical Diagnosis | |||
| IPF | 16 | 1 | 0 |
| HP | 2 | 0 | 0 |
| CTD-ILD | 1 | 0 | 0 |
| ILA | 3 | 6 | 0 |
| Self-reported medical history, n (%) | |||
| Asthma | 4 (19.0); n=21 | 2 (25.0) | 2 (25.0) |
| Ischemic heart disease | 3 (14.3); n=21 | 0 | 0 |
| Diabetes | 1 (4.8); n=21 | 1 (14.3) | 0 |
| Lung cancer | 0; n=21 | 0 | 0 |
| Hypertension | 7 (33.3); n=21 | 1 (14.3) | 3 (37.5) |
| COPD | 3 (14.3); n=21 | 0 | 0 |
| Connective tissue disease | 2 (9.5); n=21 | 0 | 0 |
| Self-reported symptoms, n (%) | |||
| Breathlessness | 16 (80.0); n=20 | 1 (14.3) | 2 (25.0) |
| Cough | 16 (80.0); n=20 | 1 (14.3) | 4 (50.0) |
| Wheeze | 8 (40.0); n=20 | 1 (14.3) | 3 (37.5) |
| Sputum | 10 (50.0); n=20 | 0 | 2 (25.0) |
| Haemoptysis | 1 (5.0); n=20 | 0 | 1 (12.5) |
| Specific examination features, n (%) | |||
| Crackles on examination | 15 (83.3); n=18 | 2 (28.6) | 0 |
| Wheezes on auscultation | 0; n=18 | 0 | 1 (12.5) |
| Clubbing | 2 (11.1); n=18 | 0 | 0 |
| Specific HRCT findings, n (%)* | |||
| Honeycombing | 11 (50.0) | 1 (14.2) | 0 |
| Ground-glass opacities | 9 (40.9) | 4 (57.1)) | 0 |
| Interlobular reticular opacities | 21 (95.5) | 6 (85.7) | 0 |
| Lung distortion | 18 (81.8) | 3 (42.9) | 0 |
| Traction bronchiectasis | 17 (77.3) | 3 (42.9) | 0 |
| Traction bronchiolectasis | 18 (81.8) | 3 (42.9) | 0 |
| Any bilateral findings | 22 (100.0) | 6 (85.7) | 0 |
| Pulmonary function measures | |||
| FVC % predicted | 78.8 (34.2-121.8) | 101.5 (84.3-115.6) | 98.1 (77.6-120.2); n=7 |
| FEV1 % predicted | 81.7 (43.3-124.8) | 100.3 (83.3-127.7) | 92.9 (65.7-115.5); n=7 |
| DLCO % predicted | 56.2 (19.6-114.1) | 88.2(68.4-117.9); n=6 | 102.2 (79.8-119.9); n=7 |
| SGRQ scores | |||
| SGRQ symptoms | 48.8 (0.0-83.8) | 10.0 (0.0-29.49) | 17.0 (0.0-39.0) |
| SGRQ activity | 52.2 (0.0-92.5) | 20.4 (0.0-48.5) | 13.2 (0.0-35.6) |
| SGRQ impacts | 30.9 (0.0-81.4) | 5.7 (0.0-25.9) | 3.6 (0.0-21.4) |
| SGRQ total score | 41.4 (6.2-85.2) | 12.1 (0.0-25.8) | 9.7 (0.0-28.7) |
| MUC5B rs35705950 genotype⁎⁎, n (%) | |||
| GG | 5 (23.8) | 2 (28.6) | 5 (62.5) |
| GT | 13 (61.9) | 5 (71.4) | 3 (37.5) |
| TT | 3 (14.3) | 0 | 0 |
| Total risk alleles (T) | 19 (45.2%); n=21 | 5 (35.7%) | 3 (18.8%) |
Genotypes were determined by Sanger sequencing of genomic DNA (primers available upon request).
Age = age at recruitment; IPF = idiopathic pulmonary fibrosis; HP = hypersensitivity pneumonitis; CTD-ILD = connective tissue disease ILD; ILA = interstitial lung abnormality BMI = body mass index; COPD = chronic obstructive pulmonary disease; FVC = forced vital capacity; FEV1 = forced expiratory volume; DLCO = diffusing capacity for carbon monoxide; SGRQ = St. George's Respiratory Questionnaire, MUC5B = Mucin 5B, Oligomeric Mucus/Gel-Forming gene.
All measurements are reported as mean(range), unless otherwise indicated. Predicted values (% predicted) were calculated using the Global Lung Function Initiatives for spirometry8 (FVC and FEV1) and the carbon monoxide transfer factor9 (DLCO).
A limitation of this study was that we were unable to examine possible ascertainment bias as we did not have access to AIPFR data for those who did not participate. In addition, the small sample size precluded formal statistical analysis to compare groups. Despite this, interesting trends have been observed. Relatives diagnosed with preclinical ILD tended to be older (71.6 vs 60.8 years) than the unaffected group. Consistent with a strong genetic predisposition in our familial cohort and earlier reports,3,5 the frequency of the established IPF risk variant in the MUC5B promotor (rs35705950) was 45.2% in the known patients, 35.7% in relatives with preclinical ILD, but only 18.8% in the unaffected group. Counterintuitively, the rate of ever smoking in the preclinical ILD group was only 28.6%, while 50.0% of their unaffected counterparts, and 54.5% of known patients, were ever smokers. This finding is consistent with a previous report,3 where the rate of ever smoking was lower in relatives from families with familial ILD compared to relatives of apparent sporadic patients (28% vs 52%), and may represent environmental risk modification in individuals with familial risk.
Preclinical ILD was only observed in siblings of patients, not offspring, with a prevalence of 58.3% when only considering relatives with an affected sibling. This finding is consistent with Aburto et al.4, who reported that 52.6% of siblings of apparent sporadic IPF patients had preclinical ILD, whilst only 5.6% of patient's offspring were affected. Given the mean age of the offspring group was only 47.7 years,4 compared with 59.4 years in our cohort, it is likely that some of these individuals may develop preclinical ILD in the future.
This report provides further evidence that clinical screening of first-degree relatives of ILD patients facilitates early diagnosis. Further research is required to determine appropriate screening for the offspring of ILD patients, however, we propose that the age of diagnosis for known patients in the family is an important consideration. Early diagnosis provides individuals with opportunities to adopt healthy lifestyle interventions, and access to improved clinical management as well as innovative treatments as they are developed.
Ethics approvalThis study was conducted in accordance with the amended Declaration of Helsinki. The study was approved by the Sydney Local Health District Human Research Ethics Committee (X18-0193), University of Tasmania Health and Medical Human Research Ethics Committee (17775) and the Prince Charles Hospital Human Research Ethics Committee (53369).
Sources of supportThis research was supported by an untied grant from a philanthropic family.
We are greatly indebted to our participants and their families for volunteering their time and clinical data; without their participation, this study would not be possible. We also thank the ILD nurses and coordinators, including Catriona Doran from Royal Adelaide Hospital; Elizabeth Ray, Shannon Cleary, Jessica Rhodes and Qi Lin from Royal Prince Alfred Hospital; Amy Cashmore from John Hunter Hospital; Sandra Bancroft from The Prince Charles Hospital; Karen Symons from The Alfred; Shona Knights-Rennie from Border Physicians Group; Samantha Wadham and Helen Rodgers from Sunshine Coast University Hospital and; David Clark from PathWest Albany. The Australian IPF Registry is facilitated by Lung Foundation Australia and is supported by unrestricted educational grants from Foundation Partners, Boehringer Ingelheim and Roche Products, Pty. Limited. SEML, PR, LT, DAS, RWB, SW, YM, SM, HW, DC, TJC and JLD made substantial contributions to the conception and design of the study; SL, KR, SM, TJC, LT, IG, PNR, DC, CC, CG, JR and PK made substantial contributions to the acquisition of patients and clinical data; SEML and JM conducted data analysis; SEML; KR; JM and TLC and JLD made substantial contributions to the interpretation of the data; SEML and JLD drafted the manuscript; while all authors were involved in revising the manuscript critically for important intellectual content; gave final approval for publishing; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SEML, TJC and JLD are guarantors of this work, taking responsibility for the integrity of the work as a whole, from inception to published article.


