




The number of bronchiectasis diagnoses has increased in the last two decades due to several factors.
Research carried out over the last years showed that an aetiological diagnosis could change the approach and treatment of a relevant percentage of patients and consequently the prognosis.
Currently, systematic investigation into aetiology, particularly of those disorders that can be subject to specific treatment, is recommended.
Given the complexity of the aetiological diagnosis, the Pulmonology Portuguese Society Bronchiectasis Study Group assembled a working group which prepared a document to guide and standardize the aetiologic investigation based on available literature and its own expertise. The goal is to facilitate the investigation, rationalize resources and improve the delivery of care, quality of life and prognosis of patients with bronchiectasis.
Bronchiectasis (BE) is characterized by irreversible airways dilatation and lesion associated with a vicious cycle of inflammation, recurrent infection and progressive bronchial damage.
In the last few years there have been many signs of a renewed interest in this pathology due to its prevalence which had not decreased in response to a better control of respiratory infections as would be expected. This fact is mainly due to a better diagnostic capacity, the recognition of the association between BE and some systemic pathologies and also to the increase in the population survival rates, namely chronic patients. Therefore, BE epidemiological characteristics have been changing, with less post-infectious BE, more adult patients and more cases associated with other prevalent diseases like chronic obstructive lung disease (COPD).1,2
BE can result from several local or systemic inherited or acquired diseases. In a significant number of cases aetiology cannot be defined, even after an extensive evaluation. Therefore, the percentage of diagnoses obtained varies widely in published series.3–5
Obtaining an aetiologic diagnosis is often considered unnecessary and non-cost effective.6 However, there are several reasons why one should investigate aetiology:
- •
The specific treatment of certain diseases causing BE relates to a better prognosis.3,4,7
- •
The presence of BE worsens the underlying disease prognosis, accelerates loss of pulmonary function, increases mortality and significantly reduces quality of life.8,9
- •
The diagnosis of hereditary diseases is important to assess transmission risk and offer genetic counselling
- •
BE is a complex, heterogeneous and possibly a multifactorial entity.10
- •
Some drugs used in cystic fibrosis (CF) are ineffective, even harmful, in non-cystic fibrosis BE,11 thus aetiology should be considered in the analysis of clinical trials because it may influence final results
- •
A clearly defined idiopathic BE patients group will help underlying mechanisms investigations
In conclusion, after diagnosing BE a systematic aetiologic investigation should be undertaken.
The Pulmonology Portuguese Society Bronchiectasis Study Group convened a working group which prepared a document to guide and standardize the aetiologic investigation based on available literature and its own experience. The goal is to rationalize resources and improve the delivery of care, quality of life and prognosis of patients with BE.
Aetiological investigationsA systematic investigation of the underlying aetiology undertaken according to a predetermined algorithm increases the likelihood of obtaining a diagnosis with better resource management.
In order to establish the most likely hypotheses and carry out the most effective tests, the initial assessment must be guided by a detailed clinical history and physical examination.
Clinical historyClinical history should include onset age, presenting symptoms, clinical evolution, previously diagnosed diseases, risk exposures, infertility history, non-respiratory symptoms and family history including consanguinity data (Table 1). These questions should be asked in a systematic way as patients may not value certain clinical data either because they do not significantly compromise their quality of life or because they are not aware of their relevance for the diagnosis.
Clinical characteristics and aetiology.
| Aetiologic clinical history | Disease |
|---|---|
| Symptoms onset age Neonatal period childhood Adult life (>40 years) Old women | PCD, CF CF Idiopathic BE NTM |
| Chronic symptoms since onset | Idiopathic BE |
| Smoking history, occupational exposures | COPD, toxic inhalation, NTM |
| Infection history | Measles, whooping cough, pneumonia, PT |
| Chronic sinusitis | PCD, CF, Young's syndrome, DPB, yellow nail syndrome, idiopathic BE |
| Nasal polyps | CF, asthma |
| Recurrent otitis media; hearing loss | PCD |
| Subfertility and infertility | PCD, CF/CFTR-RD, Young's syndrome |
| Susceptibility to infection (in other organs) | Immunodeficiencies (primary or secondary) |
| Gastrointestinal symptoms Diarrhoea Malabsortion symptoms Chronic pancreatitis/recorrent acute pancreatitis Pyrosis | IID CF CF/CFTR-RD GERD |
| Gastric band | Aspiration |
| Previously diagnosed diseases | COPD, asthma, connective tissue diseases, autoimmune diseases, IID |
| Severe asthma | ABPA |
| Family history Hereditary diseases/consanguinity | PCD, CF, Primary immunodeficiencies |
| Sputum characteristics Scarce Brown plugs | Nodular-bronchiectatic NTM–PD ABPA |
| Ethnic origin | DPB |
PCD, primary ciliary disease; CF, cystic fibrosis; DPB, diffuse panbronchiolitis; CFTR-RD, cystic fibrosis transmembrane regulator-related disease; IID, intestinal inflammatory disease; NTM-PD, non-tuberculosis mycobacteria-pulmonary disease; ABPA, aspergilosis bronchopulmonary allergic; PT, pulmonary tuberculosis.
The results of sputum microbiological examination as well as imaging features, in particular the location of BE (Fig. 1) are also data to be taken into consideration.
Aetiologic diagnosis
It is common to assume/that the diagnosis is post-infectious BE when symptoms begin after a serious infection. Nevertheless, there are cases where symptoms occur only years later when another predisposing factor is established (for example, some degree of immunodeficiency). On the other hand, one should ask the patient about respiratory symptoms arising before the infectious episode as they may be related to the first exacerbation of undiagnosed BE. Therefore, despite the fact that infectious diseases are still a frequent cause of BE,3–6,10 care should be taken in establishing the diagnosis of post-infectious BE especially in the presence of upper airway symptoms, non-respiratory symptoms or other systemic diseases previously diagnosed.
If after proper investigation aetiology is still not defined, it is important to be aware of new clinical data throughout the follow up that may require a reassessment. In some systemic diseases, such as inflammatory bowel disease and rheumatoid arthritis, BE may precede its diagnosis.12,13 In other situations, there may be an evolution over the years (for instance, a patient with IgA deficiency may progress to common variable immunodeficiency).14
In certain cases, where aetiologic diagnoses are considered of exclusion, investigation has to be rigorous.
It would be impossible to address investigation of all causes of BE in this document, so only those that currently benefit from specific monitoring and treatment strategies with prognostic implications have been included.
Alpha-1-antitrypsin deficiencyDefinitionAlpha-1-antitrypsin (AAT) is a glycoprotein, mainly produced in the liver and functions to protect the lung against proteolytic damage being a highly effective inhibitor of neutrophil elastase.
AAT deficiency (AATD) is defined by a reduced concentration of AAT in the serum and/or identification of a defective genotype. It is not a rare disease, but it is one that is underdiagnosed.15 Severe AATD affects approximately 1 in 2000 to 1 in 5000 individuals.
Protease inhibitor (PI) M is the normal allele, while the two most frequent deficient alleles are PI*S and PI*Z. The PI*Z prevalence is higher in northern and western European countries whereas the PI*S prevalence is higher in southwestern European countries. Portugal has one of the highest estimated PI*S frequency in the world (185.1/1000) with also a high PI*Z frequency (29.7/1000).16
Clinical featuresAATD is the only established genetic risk factor for COPD, and it predisposes to severe emphysema. It may also cause liver disease and, less frequently, vasculitis and panniculitis.17
The high degree of overlap between COPD and AATD is a confounder when making a clinical diagnosis. The most common pulmonary symptoms in patients with AATD include dyspnoea, cough, sputum expectoration, wheezing and frequent exacerbations.
Distinctive and suggestive features of the emphysema associated with AATD may include:
- •
Early onset (in the fourth and fifth decades)
- •
Emphysema out of proportion to risk factors (e.g. tobacco smoking, occupational exposure)
- •
Panacinar pathology
- •
Disproportionate emphysematous involvement of the lung bases
Although these suggestive features, there are no clinical characteristics that reliably distinguish COPD from AATD.
Diagnostic criteriaAATD diagnosis is based in laboratory data.
Three strategies are commonly used to diagnose AATD: measurement of the serum or plasma protein level, AAT protein phenotyping of serum or plasma, and AAT genotyping. If the AAT protein level is below the normal range, further assessment with protein phenotyping or genotyping is recommended.
As AAT is an acute phase protein, serum concentrations can be used reliably for diagnostic testing only if no inflammation is present at the time the blood sample is drawn.
A new diagnostic tool, the AlphaKit QuickScreen®, directly detects the Z protein in a sample of blood. It is a screening test (a positive indicates the need for phenotyping or genotyping) that can be performed during a clinic visit with results available in 15–30min. It has considerable potential to increase the overall AATD detection rate.
Prevalence of BE in AATDThe role of AATD in the aetiology of BE has been the subject of debate and contention over the years.18
An association was postulated after a number of case reports has linked severe (PI*ZZ phenotype) AATD to BE in individual or small numbers of cases. In these reports the incidence of BE in patients with AATD was remarkably similar, with a common overall estimate of about 40%.19–21
However, isolated case reports cannot provide any causal link between these two conditions. Moreover, some of these patients had other clinical conditions potentially leading to BE.22
A case-control study indicates that AAT phenotype distribution and gene frequencies are not different between patients with bronchiectasis and control subjects.22 However, this study highlights the importance of bronchiectasis associated emphysema. The authors show that bronchiectatic patients without emphysema do not have a different AAT distribution as compared with control subjects in contrast to bronchiectatic patients with emphysema. This suggests that bronchiectasis may be a consequence of emphysema in PI*Z patients rather than a primary cause.22
One small case-control study reported a possible increased frequency of the Z allele among patients with bronchiectasis and common variable immunodeficiency.23 Two other studies suggest that immune deficiency may combine with selected AATD phenotypes to elicit clinically significant bronchiectasis.10,24
Parr et al. have demonstrated that 27% of 74 AAT-deficient patients had High-Resolution Computed Tomography (HRCT) evidence of BE. That study indicated a concordance distribution of airway disease and emphysema with greater severity demonstrated in the lower lung. This concordance may reflect the influence of a field effect on a common pathogenic mechanism or may indicate that there is an interaction between the inflammatory processes that are considered to be putative in each condition. BE may be a consequence of emphysema, as previously suggested, but the reverse mechanism is equally plausible.25
The Canadian guidelines say that there is insufficient evidence in the literature to support routine targeted testing for AATD in individuals with BE.26
In a recent publication the REDAAT (Spanish registry of patients with AATD) group suggests that in patients with BE the AAT serum concentration should not be assessed by routine processes, but individualized.27
Further studies with a larger number of patients are required to confirm the relationship between AATD and BE and establish guidelines with a stronger evidence level.
In the context of the mixed results reported in the literature, the high frequency of deficient alleles in the Portuguese population, and the benefit from early detection of AATD (behaviour modification, optimized clinical management, existence of specific treatment for selected patients and genetic counselling) it was the consensus of the Working Group that testing for AATD should be undertaken in all individuals with BE.
AATD may play a role which is contributory to, and cumulative with other diseases as the aetiology of BE.
Recommendations- •
Screening for AATD should be made in all patients with BE.
- •
If AAT protein level is below the normal range, further assessment with protein phenotyping or genotyping is recommended.
Primary ciliary dyskinesia (PCD) is a rare, heterogeneous autosomal recessive genetic disorder with an estimated incidence of 1 per 10,000–20,000 births.28 It is characterized by abnormal ciliary ultrastructure or function, with wide clinical spectrum frequently involving recurrent respiratory infection due to defective mucociliary clearance.29 Inheritance usually follows autosomal recessive pattern.30
Clinical featuresAge at presentation range from birth to adulthood. Presenting symptoms according to age are:30–32
- •
In the newborn period
- ∘
Neonatal respiratory distress or pneumonia without any obvious predisposing factors
- ∘
Continuous rhinorrhea from the first day of life
- ∘
Associated features: disorders of laterality, complex congenital heart disease, heterotaxy, hydrocephalus, cleft palate, bilateral cervical ribs, biliary atresia, esophageal abnormalities, anal atresia
- ∘
- •
In the infants and older children
- ∘
Chronic productive cough, recurrent upper and lower respiratory tract, BE
- ∘
Chronic rhinosinusitis (rarely nasal polyps)
- ∘
Recurrent otitis media with effusion, hearing loss
- ∘
- •
In the adults
- ∘
Same as for children
- ∘
Nasal polyposis and halitosis
- ∘
Ectopic pregnancy, subfertility in females; infertility in males (50%)
- ∘
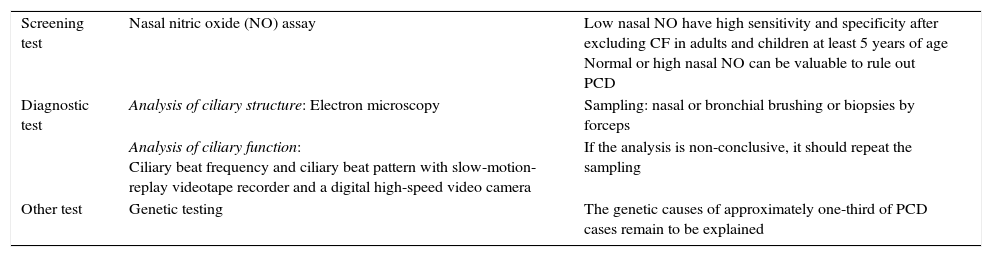
In practice, patients with clinical picture suggestive of PCD should be screened, and diseases with similar features (e.g. CF, immunodeficiency) excluded. Any positive screening testing requires confirmatory diagnostic testing to demonstrate abnormal structure and/or function of cilia.30 The screening test and the diagnostic test should be performed on clinically stable patients, free of acute upper or lower respiratory illness for at least 4–6 weeks before undergoing measurements (Table 2).30,32–34
Screening and diagnostic test for PCD.
| Screening test | Nasal nitric oxide (NO) assay | Low nasal NO have high sensitivity and specificity after excluding CF in adults and children at least 5 years of age Normal or high nasal NO can be valuable to rule out PCD |
| Diagnostic test | Analysis of ciliary structure: Electron microscopy | Sampling: nasal or bronchial brushing or biopsies by forceps |
| Analysis of ciliary function: Ciliary beat frequency and ciliary beat pattern with slow-motion-replay videotape recorder and a digital high-speed video camera | If the analysis is non-conclusive, it should repeat the sampling | |
| Other test | Genetic testing | The genetic causes of approximately one-third of PCD cases remain to be explained |
A lower threshold for screening should be considered in newborn and infants or when patient has a consanguineous background.
In adults the diagnosis of PCD should be questioned in the absence of BE.28,31
Prevalence of BE in PCDIn a cohort study 98% of adults and 61% of children with primary ciliary dyskinesia had BE.35
Recommendations- •
Patients with BE and clinical features suggestive of PCD should be screened with nasal NO assay according to standardized protocol.
- •
Electron microscopy ciliary structure analysis or video recording ciliary function analysis should be used to confirm the diagnosis.
- •
Genetic testing should not be part of the initial diagnostic testing.
Allergic Bronchopulmonary Aspergillosis (ABPA) is a non-invasive disorder caused by hypersensitivity to a ubiquitous fungus called Aspergillus fumigatus after inhalation of its spores.36 ABPA almost always occurs in patients with asthma or CF.36 It is estimated that 2% asthmatics and 1–15% of CF patients may develop ABPA but this condition remains underdiagnosed and the mean diagnostic latency is 10 years after the onset of symptoms.37
ABPA occasionally could complicate other chronic lung diseases, namely idiopathic BE or secondary to other causes.37
Clinical featuresThere is no gender predilection and the majority of cases occur in the 3rd and 4th decade.
Most patients present with low-grade fever, weight loss, worsening wheezing, worsening dyspnoea and productive cough with expectoration of very thick mucus and sometimes well-formed brownish-black mucus plugs.37,38
ABPA clinical course may evolve into 5 different Stages: Stage I – Active ABPA; Stage II – Remission; Stage III – Recurrence or exacerbation; Stage IV – Steroid-dependent ABPA; Stage V – Fibro-cavitary disease.36
Diagnostic criteriaLaboratory testsTotal serum IgE levels >1000IU/mL are the hallmark of ABPA38 (except in patients already on systemic corticosteroids) and are also the most useful test for follow-up. A doubling of patient's baseline IgE levels indicate relapse of ABPA.37
A. fumigatus-specific IgE levels are also elevated and recently were considered more sensitive than skin-prick test to Aspergillus antigen.39,40 The ideal is to use a combination of both.39
Peripheral eosinophilia (>1000cells/μL), precipitins and other IgG antibodies against Aspergillus are also supportive of diagnosis.38
ImagingHRCT demonstrates central BE (varicose or cystic) in multiple lobes – ABPA with Central Bronchiectasis (ABPA-CB), although they could be absent in some patients – Serological ABPA (ABPA-S).38 HRCT may also show mucus plugging with gloved-finger shadows (represent mucoid impaction in dilated bronchi), transient consolidation, centrilobular nodules with tree-in-bud appearance, atelectasis, mosaic perfusion (areas of air-trapping on expiration) and fibrosis (in end-stage disease).37,38
Sputum cultures for A. fumigatusSputum cultures for A. fumigatus are supportive but not diagnostic.37
There is not a single test to diagnose ABPA nor is there a universally established set of criteria,38 but the following are the recently proposed Revised Criteria:41
Obligatory criteria (all mandatory):- •
Asthma or CF
- •
Elevated total serum IgE (>1000IU/mL)
- •
Elevated Aspergillus-specific IgE levels or skin-prick test positive to Aspergillus
- •
Presence of precipitins or IgG antibodies against A. fumigatus in serum
- •
Peripheral eosinophilia (>500–1000cell/μL) in steroid-naïve patients
- •
Radiographic findings consistent with ABPA:
- ∘
Transient (consolidations; centrilobular nodules; finger-in-glove opacities)
- ∘
Permanent (central BE; pleuropulmonary fibrosis)
If the patient meets all other major and minor criteria, an IgE value<1000IU/mL may be acceptable.
Prevalence of BE in ABPAThe prevalence of BE in ABPA ranges from 69 to 78% in different studies.42
Recommendations- •
All patients with BE should be screened for sensitization to A. fumigatus with specific IgE antibody or skin-prick test.
- •
If one of the above mentioned screen tests is positive, total IgE level should be measured.
- •
A total IgE>1000IU/mL or in its absence a worsening of dyspnoea, wheezing or of productive cough should prompt the remaining diagnostic tests.
CF is the most common autosomal recessive disease among caucasian populations.43,44
It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, that results in dysfunction of the apical membrane CFTR protein, which regulates chloride and sodium transport in secretory epithelial cells, leading to abnormal ion concentrations across the apical membranes of these cells.45
CF is a multissistemic disease. It is associated to a large number of mutations (>2000),46 responsible for different types of dysfunction of the CFTR protein and consequently with a wide variability of fenotipic expression.47
The term ‘CFTR-related disorders’ is used to describe a clinical entity which is also associated with CFTR dysfunction, but that does not fulfil all the diagnostic criteria for CF.48 Three main clinical entities illustrate these phenotypes:
- •
CBAVD (congenital bilateral absence of the vas deferens)
- •
Acute recurrent or chronic pancreatitis
- •
Disseminated BE
Clinical features and their prevalence in CF patients include:49,50
- •
Disseminated BE – 95% (chronic colonization with certain microorganisms such as Staphylococcus aureus and Burkholderia cepacea complex being very suggestive of this disease)
- •
Sinus disease with panopacification of the sinuses – 90–100%
- •
Pancreatic exocrine insufficiency, leading to malabsorption – 85%
- •
Sweat gland salt loss – 100%
- •
Male infertility (absent or altered vas deferens) – 99%
- •
Meconium ileus – 20%
- •
Distal intestinal obstruction syndrome – 20%
- •
CF-related diabetes – 20%
- •
CF liver disease – 20%
- •
Nasal polyps – 10%
However, many patients demonstrate mild or atypical symptoms, with less severe pulmonary disease and pancreatic sufficiency and clinicians should remain alert to the possibility of CF even when only a few of the usual features are present.44
The clinical features of the CFTR related disorders are usually restricted to only one organ or system.
Diagnostic criteriaThe criteria to establish the diagnosis of CF are:51
- •
one or more of the characteristic phenotypic features or
- •
a history of CF in a sibling or
- •
a positive neonatal screening test result
And
- •
an increased sweat chloride concentration (>60mmol/l) or
- •
identification of two CF mutations or
- •
demonstration of abnormal nasal epithelial ion transport
As CF is associated with a more rapid progression and considerably greater mortality than non-CF BE, it is important to identify these cases.18
The diagnosis of disseminated BE with CFTR dysfunction can only be admitted in the presence of at least one CFTR mutation and after rolling out other possible aetiologies including a mild form of CF, which can be extremely difficult.48
Prevalence of BE in CFThe true prevalence of BE among children with CF is unknown; however, studies have shown that 50–70% of patients have HRCT-defined bronchiectasis by 3–5 years of age.52 Once present, BE persists and progresses in approximately 75% of young children, despite receiving of the best current therapy and by adulthood the prevalence raises to approximately 95%.
Recommendations- •
All children presenting with BE will need investigations to exclude CF.
- •
Adults presenting with BE will need investigations to exclude CF especially if the following clinical features are found:
- ∘
Gastrointestinal malabsorption
- ∘
Male primary infertility
- ∘
History of childhood steatorrhoea
- ∘
Persistent isolation of Staphylococcus aureus in sputum
- ∘
Upper lobe bronchiectasis
- ∘
- •
Screening investigations should include two measurements of sweat chloride (i.e. quantitative test) and CFTR genetic mutation analysis.
- •
In patients with BE with no obvious aetiology atypical CF and CFTR dysfunction related BE should be considered.
- •
In case of positive or borderline sweat tests patient should be referred to a specialist CF centre. The same should be done in patients with negative sweat tests but a strong clinical suspicion of CF.53
Primary immunodeficiencies (PIDs) are a heterogeneous group of rare and complex diseases that include more than 230 different disorders caused by ineffective immune function resulting from different mechanisms.54 Predominantly antibody deficiencies constitute the most prevalent group of PIDs.54,55 These include the PIDs that more frequently present with BE as sequelae of recurrent chest infections3,56,57 particularly common variable immunodeficiency (CVID), specific antibodies deficiency, X-linked or autossomic recessive agammaglobulinaemia, IgA deficiency and Good syndrome.
PID recognition is crucial for early diagnosis and appropriate access to treatment, limiting infections and secondary structural damage. Considering PID is the key to diagnosis.58
Clinical featuresThe hallmark of PID is infection. Patients with antibody deficiencies present with recurrent infections, most frequently caused by encapsulated bacteria and affecting the respiratory tract. These infections, besides increased frequency, typically fail regular treatment and become chronic or evolve to severe complications.58
CVID is the most prevalent symptomatic PID in adulthood (1:50,000 inhabitants in western countries).59 Besides infections, other features may be found in CVID, including prolonged or recurrent diarrhoea and/or different expressions of autoimmunity or lymphoid proliferation.
Specific antibody deficiencies are much more rare and are frequently associated with complicated BE. In 2008, Agamohammadi et al. reported that 30% of the patients with BE classified as idiopathic presented decreased response to vaccination, allowing their aetiologic reclassification as specific antibody deficiency.60
PID diagnosis is often missed when specific antibody testing is not performed in patients with recurrent chest infections and normal levels of total serum IgG.60
Agammaglobulinaemia is a PID characterized by the arrest of B cell differentiation, leading to a considerably reduced B lymphocyte count and very low serum Ig levels. Infections usually start occurring between 3 and 6 months of age, when maternal IgG levels start to decline. Lifelong immunoglobulin replacement therapy is indicated. Early treatment with IgG is essential to reduce the infections recurrence and severity although BE may develop during follow-up in patients with adequate IgG replacement therapy.61
IgA deficiency is the most frequent PID (1:600 inhabitants in western countries), but the vast majority of patients in this group is asymptomatic.62 If IgA deficiency is diagnosed in a patient with BE, IgG subclasses quantification and specific antibody testing must be performed.60 IgA deficiency may evolve to CVID or specific antibody deficiency.
Good syndrome (timoma with immunodeficiency) is a rare cause of combined T and B cell immunodeficiency. Usually present in the 4th or 5th decade of life and clinical characteristics are increased susceptibility to bacterial infections with encapsulated microorganisms and opportunistic viral and fungal infections.63
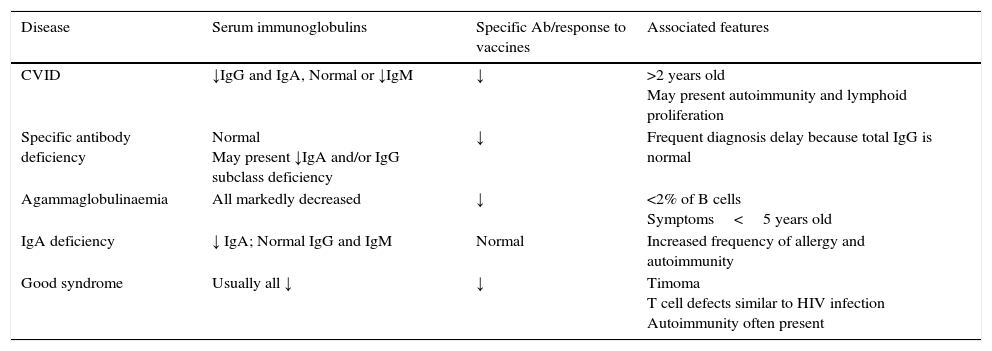
The main features of these pathologies are mentioned in Table 3. Other causes of hypogammaglobulinemia must be excluded.
PID characteristics.
| Disease | Serum immunoglobulins | Specific Ab/response to vaccines | Associated features |
|---|---|---|---|
| CVID | ↓IgG and IgA, Normal or ↓IgM | ↓ | >2 years old May present autoimmunity and lymphoid proliferation |
| Specific antibody deficiency | Normal May present ↓IgA and/or IgG subclass deficiency | ↓ | Frequent diagnosis delay because total IgG is normal |
| Agammaglobulinaemia | All markedly decreased | ↓ | <2% of B cells Symptoms<5 years old |
| IgA deficiency | ↓ IgA; Normal IgG and IgM | Normal | Increased frequency of allergy and autoimmunity |
| Good syndrome | Usually all ↓ | ↓ | Timoma T cell defects similar to HIV infection Autoimmunity often present |
Several reports have shown that IgG replacement therapy should ensure serum IgG levels (above 1000mg/dL) in order to allow a cutback in the progression of lung deterioration and decrease of severe bacterial infections frequency.64–66
Diagnostic criteria – investigation of PID- •
Serum immunoglobulin levels – IgG, IgA, IgM and IgE
- •
Serum IgG subclass titres
- •
Specific antibodies (quantification by ELISA) – total IgG and IgG2 for pneumococcal antigens and IgG1 for tetanus toxoid antigen
- ∘
if low titters are detected, access vaccine response after 4–6 weeks; specific antibodies should increase >4 times pre-vaccination level
- ∘
- •
Peripheral blood lymphocyte main subpopulations–including T cells (total CD3 and CD4 and CD8 subpopulations), B cells and NK cells
BE has been reported in up to 76% of patients with CVID, being more frequent in cohorts with a longer diagnosis delay. BE development in CVID has been associated with severe/recurrent respiratory tract infections and history of pneumonia,67 unregulated inflammation, lower numbers of memory B cells and CD4+ T-cell counts below 700/μl.68
A study of lung complications in 30 patients with antibody deficiencies showed that 53% had CT scans abnormalities. Main abnormality was BE (12/16 patients), which was seen more frequently (67%) on the right middle lobe.57
Recommendations- •
All patients with BE should have measurement of serum IgG, IgA and IgM levels and serum electrophoresis.
- •
BE found in a patient with IgA deficiency should always prompt specific antibodies production evaluation.
- •
In patients with BE with no obvious aetiology specific antibody production should be tested.
- •
The presence of BE should be assessed in all patients with PID and PID should be considered in patients with BE.
- •
Patients with PID should be referred to an immunology specialist.
Infections with nontuberculous mycobacteria (NTM) are caused by a large group of mycobacterial species, with varied spectrum of pathogenicity, other than those of the Mycobacterium tuberculosis complex and Mycobacterium leprae.
NTM are ubiquitous bacilli whose main reservoirs are water and soil. In this context exposure to these organisms is constant.69
The usual classification of NTM is established with respect to time of growth in culture.70
Slow growth of the NTM (>7 days), more pathogenic, have Mycobacterium avium complex as the most frequent cause of pulmonary NTM infection.71 Fast-growing NTM (<7 days), with a less aggressive range of organisms, can produce illness with high therapeutic difficulty and worse prognosis as Mycobacterium abscessus complex.72
Clinical featuresPulmonary involvement is the most common disease manifestation of NTM infections (70–90% of cases). The most frequent presentation is the chronic progressive form, rarely, hypersensitivity pneumonitis (hot tub lung).73,74
The chronic clinical presentation of nontuberculous mycobacterial pulmonary disease (NTM-PD) can be separated into two relevant patterns:70
- •
Patients with pre-existing structural changes characterized by BE and/or cavitations resulting of previous lung diseases, especially in elderly men.
- •
Patients with multifocal nodular BE of slow evolution and without lung underlying disease in non-smoking women (often with abnormalities of the chest cage and low body mass index).
Typical presenting symptoms of NTM-PD include respiratory symptoms such as persistent cough, often purulent sputum, sometimes haemoptysis, dyspnoea and systemic symptoms with asthenia, weight loss and intermittent fever.71
BE are associated with NTM-PD as a predisposing factor to the disease, as well as a result of it, keeping a cause and effect cycle which contributes to worsening structural damage of the airways with progressive clinical deterioration.75–77
Diagnostic criteriaGiven the relatively low virulence of the NTM, in order to produce disease, the host has usually some level of systemic or local immune deficiency.78–80
Although not yet clinically validated, ATS/IDSA proposed diagnostic criteria of NTM-PD which includes the following:81
- •
Suggestive clinical picture and imaging findings
- •
Exclusion of other similar diseases (fungal disease, malignancy, tuberculosis)
- •
Identification of NTM:
- ∘
at least two positive culture from separate sputum samples or
- ∘
culture positive in at least one sample of bronchial wash/lavage or
- ∘
biopsy showing histopathologic features (granulomatous inflammation) and positive culture in 1 sputum sample or 1 sample LBA or 1 biopsy sample
- ∘
Patients who are suspected of NTM-PD but do not meet the essential criteria should be kept under surveillance until firmly confirmation or exclusion of this diagnosis.82
EpidemiologyOverall NTM infections have increased significantly in recent years. There is however a great variability between regions regarding the prevalence and isolated species.83,84
The incidence of tuberculosis and NTM infection is inversely proportional. In this sense there has been a substantial increase in NTM disease in areas of low incidence of tuberculosis.85
Epidemiological studies indicate the following data:86,87
- •
USA – incidence of 5–6/100,000 year; prevalence 10–40/100,000
- •
Canada – prevalence 14.1/100,000
- •
Australia – incidence 3.3/100,000
- •
Great Britain – incidence 2.9/100,000
In a recent study, NTM were found in 37% of non-cystic BE, 30% of which met the NTM disease criteria.88
RecommendationsGiven the strong association of BE with NTM infection it is important to rule out NTM in the following groups of patients with BE:7,15,89
- •
In the initial evaluation.
- •
Before starting long-term use of macrolides.
- •
Every six months when on long-term use of macrolides.
- •
In the suspicion of NTM-PD (clinical deterioration with unexplained persistent respiratory and systemic symptoms and/or with new infiltrated/cavitations not responding to usual treatment with regular antibiotics).
- •
Every year when there are no specific risk factors.
NTM identification should be made:90
- •
In sputum, 3 samples, preferably taken in the morning, on different days.
- •
In BAL/biopsy when patients are paucibacillary with little or no sputum (nodular bronchiectatic type) and suspected of NTM-PD.
The presence of BE in a patient with obstructive airways disease may result from the intersection of prevalences or a causal relationship. Pathophysiologically it is acceptable to consider a causal link but no longitudinal studies have proved this relationship yet.2
Clinical featuresClinically these patients have increased systemic inflammation, poorer pulmonary function, greater number and severity of exacerbations, more production and purulence of sputum, worse quality of life and worse prognosis.2,91
In COPD, BE are typically cylindrical, small and multiple, predominating in lung bases.91 In asthma, BE can affect all lobes equally.18
Diagnostic criteriaDiagnosis of obstructive airway diseases (asthma or COPD) with BE on high-resolution CT without evidence of other cause.
Prevalence of BE in obstructive airway diseasesPrevalence of BE in patients with COPD is highly variable (2–74%),91,92 probably in relation with the criteria used to define BE. Several studies have confirmed a prevalence of approximately 50% in patients with moderate-severe COPD.91,93,94
The prevalence of BE in asthma is described between 17.5 and 28%.18
Recommendations- •
BE should be suspected in patients with obstructive airway diseases (COPD and asthma) and frequent exacerbations.
- •
If clinically relevant, other BE aetiologies should be excluded.
Diffuse panbronchiolitis (DPB) is an idiopathic inflammatory disease that mainly affects the respiratory bronchioles, causing a suppurative and progressive severe obstructive disorder. Histologically it is characterized by chronic inflammation, centralized in the respiratory bronchioles with accumulation of histiocytes, neutrophils and lymphocytes. Foamy macrophages in respiratory bronchioles wall and surrounding interalveolar septa are one feature of DPB.95
Clinical featuresDPB is most prevalent in the Asian population but there are cases and small series described in Caucasians, Hispanics and Afro-Americans.
It is usually diagnosed in the 2nd–5th decade of life and 2/3 of patients are nonsmokers.95
The first symptom is productive cough with abundant sputum, followed by progressive exertional dyspnoea. More than 80% of patients have chronic paranasal sinusitis.
The most frequently isolated pathogen in sputum is H. influenza, while infection with P. aeruginosa is common in later stages.
The most characteristic laboratory finding is the persistent elevation of cold agglutinins.
Pulmonary function tests usually show an obstructive pattern and low values of nasal nitric oxide.95,96
Chest CT shows poorly defined centrilobular nodules, signs of air-trapping and bronchial wall dilation/thickening. Changes are scattered but more significant in the lower lobes.96
Response to macrolides is excellent.
Diagnostic criteriaCases definitely established should fulfil criteria 1, 2 and 3, along with at least two of criteria 4, 5 and 6.95
- •
Persistent cough, sputum and exertional dyspnoea
- •
History of chronic paranasal sinusitis
- •
Bilateral diffuse small nodular shadows on a plain chest radiography film or centrilobular micronodules on chest computed tomography images
- •
Coarse crackles
- •
FEV1/FVC <70% and PaO2<80mmHg
- •
Titre of cold haemagglutinin≥64
- •
Due to its rarity, in Caucasians diagnosis should be based on lung biopsy.
The prevalence of BE in PBD is 100% in advanced stages.96
Recommendations- •
In a Caucasian patient with clinical criteria, lung biopsy should be considered.
Gastro-oesophageal reflux disease (GERD) is a pathological condition which develops when gastric reflux causes quality of life disturbing/troublesome symptoms and/or local complications.
Clinical featuresAccordingly to the Montreal Consensus Conference,97 GERD manifestations are grouped in two big syndromes: esophageal and extraesophageal (Fig. 2):

Concerning esophageal syndromes, symptomatic syndromes can be considered in an initial evaluation. They include typical GERD symptoms (retroesternal burning and acid regurgitation) and chest pain. An endoscopic evaluation with biopsy will determine if there is lesion of the esophageal mucosa – syndromes with esophageal injury (Fig. 2).
Within extraesophageal syndromes two subgroups were considered accordingly to the causal links observed: established association and proposed association (Fig. 2).
In these syndromes, the pulmonary disorders where reflux is demonstrated include cough and reflux asthma. In idiopathic pulmonar fibrosis reflux is only considered a probable association.
Other reflux/GERD associated pulmonary diseases are referred to in the literature98,99 but the causal relationship is not established with the same probability degree as to the listed above. Among them are:
- •
Pulmonary aspiration complications (lung abscess, BE, aspiration pneumonitis)
- •
Chronic bronchitis
- •
COPD
- •
Obstructive sleep apnea syndrome
The causal relationship between GERD and BE is controversial, especially in adults, but it is recognized that GERD could have a negative clinical impact on BE patients. Some studies demonstrated an association with a poorer lung function and a more extensive disease on HRCT.100
Physiopathological mechanisms that can explain GERD pulmonary manifestations are:101,102
- •
Vagal reflex mechanisms with bronchospasm resulting of vagal stimulation secondary to esophageal acid content (reflux asthma syndrome)
- •
Stimulation of esophageal vagus nerve endings resulting in reflex cough (reflux cough syndrome)
- •
Microaspiration/gastric reflux to the airways (mechanism proposed mainly on reflux cough syndrome and not so much on reflux asthma syndrome). It is known that the gastric content can be acidic or gaseous.
Extraesophageal syndromes show distinctive features from esophageal syndromes with reference to antireflux therapy response and multifactorial causes being esophageal reflux only a cofactor.
Diagnostic criteriaDiagnostic and therapeutic approach of extraesophageal syndromes associated GERD (namely pulmonary):99,101
- •
In patients with typical GERD symptoms a therapeutic empiric trial could be done:
- ∘
double dose proton pump inhibitors (PPI) 30min before breakfast and dinner for 12 weeks (ex: esomeprazole 40mg; pantoprazole 40mg; lansoprazole 30mg).
- ∘
if symptoms improve and the patient is well after the 12 weeks trial, extraesophageal syndrome can be attributed to GERD; PPI dose should be reduced and evolution assessed.
- ∘
- •
In patients with GERD atypical symptoms 24-h oesophageal pH-testing with impedance study should be performed. If the exam confirms the diagnosis, therapeutic trial can be done. If not, an alternative cause for pulmonary symptoms other than GERD must be considered.
In a study by Pasteur et al. considering several BE aetiological factors, GERD was identified as causative factor in 4% of patients.5
The reported GERD prevalence in the Western world ranges between 10 and 20%.103
Recently, an observational prospective study, conducted to determine the prevalence of proximal and distal gastroesophageal reflux in COPD and BE patients, showed that it was 2 times higher when compared to a control population. The prevalence of GERD in BE was 40%, of whom 42% had clinically silent reflux.104
Recommendations- •
GERD should be suspected in the presence of typical symptoms or patients with poorly controlled respiratory symptoms.
The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.


