








Short telomeres are recognized as risk factor for idiopathic pulmonary fibrosis (IPF). We aimed to assess the role of telomere length (TL) in fibrotic-Interstitial Lung Diseases (f-ILDs) associated with a usual interstitial pneumonia (UIP) pattern as well as in IPF acute exacerbation (IPF-AE).
Aim and methodsTL was measured from peripheral white blood cells using a multiplex quantitative polymerase chain reaction in consecutive patients with f-ILDs, all presenting UIP pattern in the high-resolution chest-computed-tomography and compared to age-matched healthy controls.
ResultsSeventy-nine individuals were included (mean age 69.77 ± 0.72 years); 24 stable IPF, 18 IPF-AE, 10 combined pulmonary fibrosis and emphysema, 7 Rheumatoid arthritis-UIP-ILDs and 20 controls. TL in all patients was significantly shorter compared to controls [mean T/S ratio (SE) 0.77 (±0.05) vs 2.26 (±0.36), p < 0.001] as well as separately in each one of f-ILD subgroups. IPF-AE patients presented significantly shorter TL compared to stable IPF (p = 0.029). Patients with IPF and shorter than the median TL (0−0.72) showed reduced overall survival (p = 0.004). T/S < 0.72 was associated with increased risk for IPF-AE (OR = 30.787, 95% CI: 2.153, 440.183, p = 0.012) independent of age, gender, smoking and lung function impairment. A protective effect of TL was observed, as it was inversely associated with risk of death both in UIP-f-ILDs (HR = 0.174, 95%CI: 0.036, 0.846, p = 0.030) and IPF patients (HR = 0.096, 95%CI: 0.011, 0.849, p = 0.035).
ConclusionsShorter TL characterizes different UIP f-ILDs. Although no difference was observed in TL among diverse UIP subgroups, IPF-AE presented shorter TL compared to stable IPF. Reduced overall survival and higher hazard ratio of death are associated with shorter TL in IPF.
Fibrotic interstitial lung disease (f-ILD) is the end result of a wide spectrum of rare diseases that affect the pulmonary interstitium. In this heterogeneous group, idiopathic interstitial pneumonias (IIPs), connective tissue associated-ILD as well as combined pulmonary fibrosis and emphysema (CPFE) are included.1–3 Telomeres are DNA–protein structures that protect chromosome ends and preserve genetic information. In each cell division telomeres shorten which leads finally to apoptosis and cell cycle arrest when reaching a critical point.4,5 The role of telomerase is crucial, a specialized polymerase responsible for restoring the telomere loss and protecting TL by adding telomere repeats - six nucleotides (TTAGGG) - to the ends of chromosomes.6–8 Telomerase consists of two essential components: the telomerase reverse transcriptase (hTERT), a catalytic component, and an RNA component (hTR), the template for nucleotide addition by hTERT.6–8 In patients with familial pulmonary fibrosis, mutations in either of the essential components of telomerase can be found leading to acceleration of telomere shortening.9 Furthermore, it has been shown that patients with IIPs who do not carry the above mutations, have shorter telomeres, compared to healthy controls.10
Idiopathic pulmonary fibrosis (IPF) which is histologically characterized by the usual interstitial pneumonia pattern (UIP), represents the most prevalent among IIPs, carries the worst prognosis,11,12 and is the better characterized telomere-mediated-associated ILD.6–8,13–20 As a matter of fact, in previous studies peripheral blood telomere length (TL) has been found to be shorter in IPF patients compared to age-matched controls and thus nowadays, short telomeres constitute a well-defined risk factor for IPF.6–8,13–20 Recently, the association of short telomeres with other ILDs, such as Rheumatoid Arthritis-UIP-ILD (RA-UIP-ILD) has also emerged.21–23 There is still lack of evidence concerning TL in CPFE as well as IPF acute exacerbation (IPF-AE), both originally characterized by the common denominator of UIP histology. So far, to the best of our knowledge, no study has examined the association of TL across different UIP fibrotic-ILDs.
IPF patients may experience a catastrophic event representing the development of diffuse alveolar damage (DAD) upon UIP called IPF-AEs.24–26At present, the pathogenesis of such events is attributed to several triggering factors including infection, aspiration, and gastroesophageal reflux with or without the additional effect of air pollution.24–27 So far, to the best of our knowledge, the potential association of TL with the appearance of IPF-AE has not been investigated despite the evidence that short telomeres are associated with limited lung tissue renewal capacity.4,9
The aim of the present study is to assess TL in Greek patients with sporadic fibrotic ILDs associated with UIP pattern, including IPF, IPF-AE, RA-UIP-ILD and CPFE and compare it with healthy controls.
Material and methodsStudy subjectsConsecutive patients with sporadic fibrotic ILD, such as IPF, IPF-AE, RA-UIP- ILD and CPFE who presented a UIP pattern in the HRCT and with no family history of pulmonary fibrosis, referred to our department from October 2016 until November 2017 were included in this prospective study as cases. Disease diagnosis was according to international guidelines after applying a multidisciplinary approach for each case.11,12 Special care was attributed to the clinical history of each patient using the questionnaire developed by the American College of Chest Physicians (CHEST) to uncover any potential exposure source and exclude chronic hypersensitivity pneumonitis cases.28,29 Furthermore, the definite UIP pattern on HRCT was confirmed by the presence of honeycombing, traction bronchiectasis and bronchiolectasis after being evaluated by two independent readers (SA, SAP).12 Patients with IPF-AE, fulfilled the proposed criteria of the international consensus, including IPF diagnosis, worsening of dyspnea within 30 days of an unidentifiable cause and not fully explained by cardiac failure, while any new ground glass opacities or consolidations were confirmed by chest computed tomography (CT) scan at the time of admission.11,24 All patients were followed-up regularly every three months. Finally, 20 age-matched healthy individuals visiting hospitalized patients at the same time as the ILD patients in our department were selected as controls. The study protocol was approved by the local Ethics Committee (EBΔ 201/23-4-14) and all participants provided written informed consent.
Study designInformation on demographics, medical history and smoking status were collected for all participants. Moreover, all participants were submitted to physical examination. Information about patients, clinical and laboratory findings including pulmonary function tests, time since first diagnosis, comorbidities as well as data on any immunosuppressive and/or antifibrotic treatment were recorded. All patients hospitalized for acute exacerbation, had computed tomography pulmonary angiogram (CTPA) performed to exclude pulmonary embolism. Also, in an attempt to reveal any pathogens, C - reactive protein (CRP) was measured and blood, sputum and bronchoalveolar lavage (BAL) cultures, where feasible, were performed.
Blood collection-DNA extractionA blood sample of 5 mL was collected from participants using the standardized phlebotomy. Directly after the collection, blood samples were used for DNA extraction, using the QIAamp DNA Blood Mini Kit (Qiagen, Heidelberg Germany). DNA concentration was measured by QIAexpert (Qiagen, Heidelberg Germany) and only good-quality DNA with an A260/A280 ratio of 1.7–2.0 stored long-term in TE at −20 °C for further experimental procedures.
Telomere length measurementTelomere length of genomic DNA from circulating leukocytes was determined using a multiplex quantitative polymerase chain reaction (qPCR) method as described by Cawthon30 which is the predominant method used to measure average telomere length. The final concentrations of reagents and the thermal cycling conditions in the qPCR were as described by Cawton.30 Serial dilutions of a reference DNA sample from the control group served as the “standard DNA” and was run in triplicates to generate the standard curves used for relative quantitation. All experimental DNAs were assayed in triplicate. This method is an extension of basic PCR whereby amplification and relative quantification of target DNA occurs simultaneously. This is achieved by coupling the target DNA with a fluorescent dye, where the amount of fluorescence generated is proportional to the amount of product. Relative quantification is calculated by dividing the telomeric DNA product (T) by the reference gene (S) that is present as a single copy in the genome, to generate a (T/S ratio). All the experiments were carried out at the Rotor-Gene Real Time PCR cycler (Qiagen, Heidelberg Germany).
Statistical analysisContinuous variable (age, telomere length etc.) differences among disease groups and controls were evaluated using non-parametric testing (Mann-Whitney U test for two groups, Kruskal–Wallis for more than 2). For interval/ordinal variables chi square was performed. Kaplan Meyer was used for survival analysis and a binary logistic regression model with an outcome of exacerbation in IPF patients controlling for potential confounders. All p reported were two-sided with statistically significant results reported when p < 0.05. Statistical analysis was performed using SPSS 25 (IBM SPSS Statistics for Windows, Version 25.0. Armonk, NY: IBM Corp. Released 2017).
ResultsDemographic and clinical characteristics of the study subjects are presented in Table 1. In total, 79 participants were included in the study (mean age 69.77 ± 0.72 years). More specifically, 24 stable IPF (70.2 ± 1.3 years), 18 IPF-AE (71.2 ± 1.7 years), 10 CPFE (70.9 ± 1.4 years), 7 RA-UIP-ILD (68.9 ± 3.5 years) and 20 age-matched healthy controls (67.8 ± 1.2 years) were enrolled. In our cohort the majority of individuals were males in all studied subgroups except RA-UIP-ILD. The distribution of gender did not significantly differ between the total number of patients with a UIP pattern and controls (p΄-value = 0.583) (Table 1). Among IPF patients, those with IPF-AE had significantly lower diffusing capacity of the lung for carbon monoxide percent (DLCO %) and forced vital capacity percent (FVC %) values but higher CRP (p = 0.058, p = 0.003 and p = 0.042, respectively). None of the patients received immunosuppressive therapy during IPF-AE whilst all received broad-spectrum antimicrobial coverage (data not shown). Also, CPFE patients had significantly lower DLCO % than IPF stable ones (p = 0.015).
Patients’ demographic, functional, clinical and laboratory characteristics n = 79.
| IPF stable | IPF-AE | CPFE | RA-UIP-ILD | Controls | p-value | UIP pattern overall | p΄-value | |
|---|---|---|---|---|---|---|---|---|
| n=79 | 24 | 18 | 10 | 7 | 20 | 59 | ||
| Age, years- mean (SE) | 70.2 (± 1.3) | 71.2 (± 1.7) | 70.9 (±1.4) | 68.9 (±3.5) | 67.8 (±1.2) | 0.477 | 70.4 (±0.9) | 0.082 |
| Male -n (%) | 16 (66.7) | 14 (77.8) | 10 (100) | 1 (14.3) | 12 (60) | 0.002 | 41 (69.5) | 0.583 |
| DLCO %pred-mean (SE) | 53.5 (± 3.8) | 38.7 (± 4.0) | 34.0 (± 5.22) | 42.0 (± 6.84) | N/A | 0.009* | (43.7 ± 2.49) | N/A |
| FVC %pred-mean (SE) | 83.7 (± 4.3) | 60.2 (± 4.3) | 81.7 (± 4.4) | 72.6 (± 5.5) | N/A | 0.003** | 73.9 (±2.76) | N/A |
| LTOT-n (%) | 1 (4.2) | 10 (55.6) | 5 (50.0) | 1 (14.3) | 0 (0) | <0.001 | 17 (28.8) | 0.004 |
| CRP, mg/dL- mean (SE) | 5 (± 2) | 47 (± 26) | 23 (± 18) | 21 (± 8) | N/A | 0.039*** | 24.9 (±7.1) | N/A |
Abbreviations: IPF: Idiopathic Pulmonary Fibrosis, IPF-AE: Idiopathic Pulmonary fibrosis Acute Exacerbation, CPFE: Combined Pulmonary Fibrosis and Emphysema, RA-UIP-ILD: Rheumatoid Arthritis-usual interstitial pneumonia associated Interstitial Lung Disease, UIP: Usual Interstitial Pneumonia, FVC: Forced Vital Capacity, DLCO: Diffusing Capacity of the lung for carbon monoxide, LTOT: long-term oxygen therapy, CRP: C-reactive protein. Bold represent statistically significant differences at p<0.05.
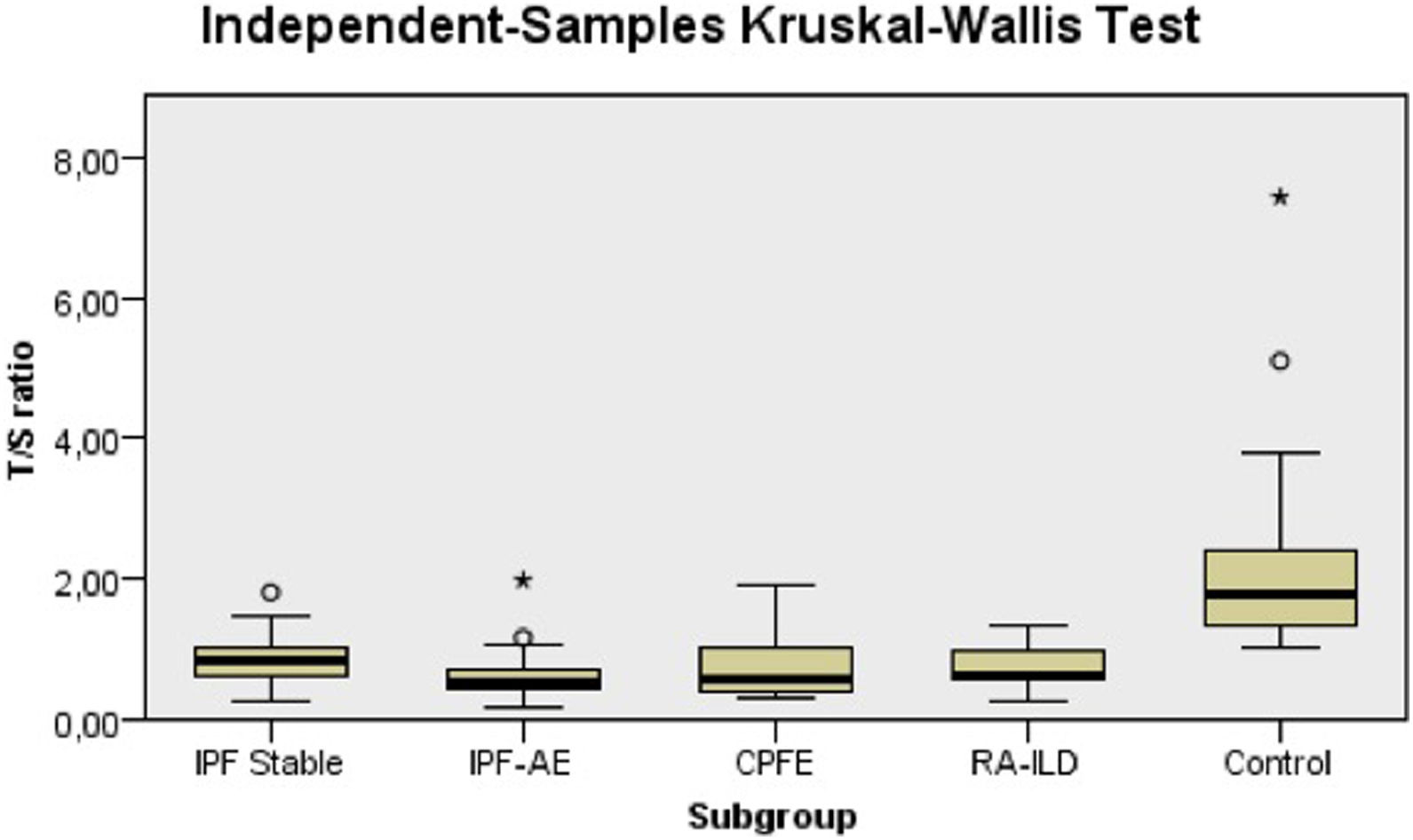
Telomere length detected in peripheral blood leukocytes of patients was significantly shorter compared to controls [mean T/S ratio (SE) 0.77 (±0.05) vs 2.26 (±0.36), p < 0.001] (Table 2). Also, shorter TL was observed in each one of the subgroups examined compared to the healthy controls [mean T/S ratio (SE) 0.87 (±0.07) in IPF stable vs. 0.66 (±0.10) in IPF-AE vs. 0.77 (±0,16) in CPFE vs. 0.74 (±0.15) in RA-UIP-ILD vs. 2.26 (±0.36) in controls, p < 0.001] (Table 2, Fig. 1). When we compared TL among the diverse subgroups no difference was observed except for patients with IPF-AE who had shorter TL compared to patients with stable IPF [mean T/S ratio (SE) 0.87 (±0.07) vs 0.66 (±0.10), p = 0.029]. Using a cut-off of 0.72, that is the median value of all IPF cases, patients with IPF-AE were significantly more frequently identified in the lower TL group (77.8% vs. 29.2% in stable IPF, p = 0.004).
Comparison of telomere length between ILD patients presenting with a UIP pattern (subgroups and overall) and controls.
| IPF stable | IPF-AE | CPFE | RA-UIP-ILD | Controls | p-value | UIP pattern overall | p΄-value* | |
|---|---|---|---|---|---|---|---|---|
| n=79 | 24 | 18 | 10 | 7 | 20 | 59 | ||
| T/S- mean (SE) | 0.87 (±0.07) | 0.66 (±0.10) | 0.77 (±0,16) | 0.74 (±0.15) | 2.26 (±0.36) | <0.001 | 0.77 (±0.05) | <0.001 |
Abbreviations: IPF: Idiopathic Pulmonary Fibrosis, IPF-AE: Idiopathic Pulmonary fibrosis Acute Exacerbation, CPFE: Combined Pulmonary Fibrosis and Emphysema, RA-UIP-ILD: Rheumatoid Arthritis-usual interstitial pneumonia associated Interstitial Lung Disease, UIP: Usual Interstitial Pneumonia. Bold represent statistically significant differences at p < 0.05.
Using the same median cut-off, as mentioned above, patients were grouped in low TL and high TL. Idiopathic pulmonary fibrosis patients with lower TL (<0.72) showed reduced overall survival than those with a longer one (13.26 ± 1.62 months vs 29.7 ± 5.6, p = 0.004) (Fig. 2). This was not the case in CPFE (p = 0.285) or RA-UIP-ILD (p = 0.527). In Table 3 the results of cox univariate and multivariate regression analysis are presented. The association of T/S ratio (treated as a continuous variable) with the risk of death in UIP f-ILDs as well as in the IPF patients remained significant after adjusting for several confounders, lung function included (HR: 0.174, 95%CI: 0.036-0.846, p = 0.030 and HR: 0.096, 95%CI: 0.011−0.849, p = 0.035, respectively). To be precise, a 0.01 increase of TL length is associated with a 17.4% and a 9.6% decrease in the risk of death in UIP f-ILD and IPF patients respectively. Acute exacerbation of IPF had the most profound effect on overall survival (14.7 ± 3.55 months vs. 23.7 ± 1.38 months, p < 0.001). Using a binary logistic regression model we found a significant association of TL with IPF-AE (OR = 8.5, 95%CI: 2.06–35.08, p = 0.003), a finding independent of age (p = 0.636), gender (p = 0.433) and smoking (p = 0.499). This association remained significant (p = 0.020) after controlling for years from diagnosis, LTOT, DLCO, FVC, parameters that could reflect a more severe disease (Table 4).
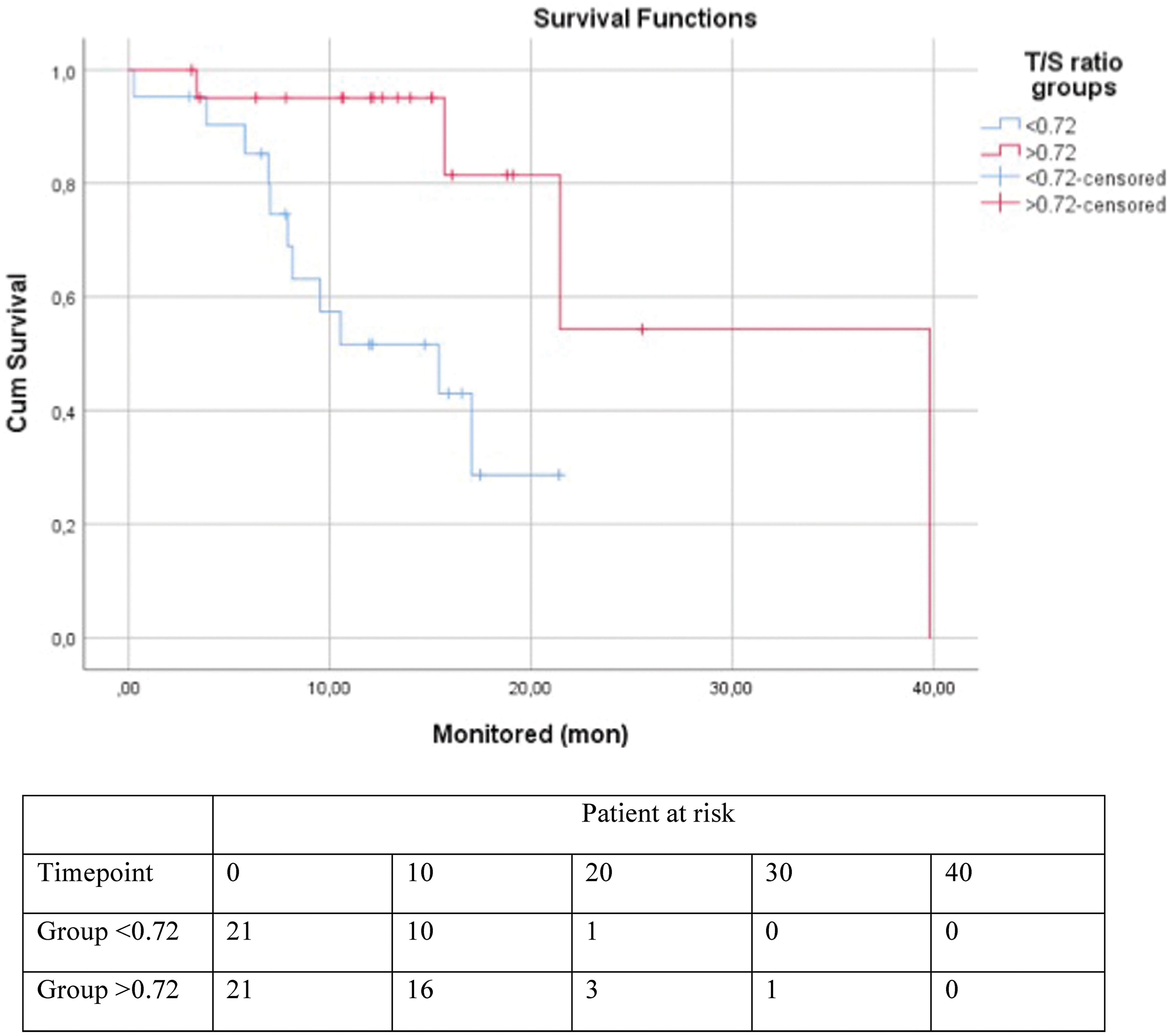
Risk of death in UIP f-ILDs patients.
| Cox Regression | Parameters | HR | (95% CI) | p-value |
|---|---|---|---|---|
| Univariate Analysis | ||||
| T/S ratio groups (Indicator: TL > median value)a | 3.722 | (1.233, 11.231) | 0.020 | |
| T/S ratiob | 0.149 | (0.034, 0.653) | 0.012 | |
| Age | 0.990 | (0.925, 1.058) | 0.762 | |
| Sex (Indicator: Male) | 2.268 | (0.755, 6.808) | 0.144 | |
| Subgroup (Indicator: IPF stable) | 0.022 | |||
| IPF-AE | 10.092 | (2.236, 45.550) | 0.003 | |
| CPFE | 9.5494 | (1.698, 54.202) | 0.001 | |
| RA-UIP-ILD | 4.733 | (0.665, 33.682) | 0.121 | |
| Years from Diagnosis | 0.031 | (1.015, 1.368) | 0.031 | |
| LTOT | 3.007 | (1.233, 7.331) | 0.015 | |
| CRP | 1.006 | (1.000, 1.011) | 0.033 | |
| pCT | 48.589 | (2.710, 871.205) | 0.008 | |
| DLCO | 0.961 | (0.924, 0.999) | 0.044 | |
| FVC | 0.978 | (0.953, 1.004) | 0.093 | |
| Multivariate Analysis (Results only for T/S ratio presented) | ||||
| T/S ratio + Age | 0.133 | (0.028, 0.618) | 0.010 | |
| T/S ratio + Sex | 0.153 | (0.034, 0.694) | 0.015 | |
| T/S ratio + Subgroup (IPF stable, IPF-AE, CPFE, RA-UIP-ILD) | 0.224 | (0.055, 0.910) | 0.036 | |
| T/S ratio + Years from Diagnosis | 0.210 | (0.051, 0.872) | 0.032 | |
| T/S ratio + LTOT | 0.128 | (0.029, 0.566) | 0.007 | |
| T/S ratio + CRP | 0.171 | (0.043, 0.680) | 0.012 | |
| T/S ratio + pCT | 0.050 | (0.003, 0.867) | 0.040 | |
| T/S ratio + DLCO | 0.205 | (0.046, 0.905) | 0.037 | |
| T/S ratio + Years from Diagnosis + LTOT + CRP + pCT + DLCO | 0.001 | (0.000, 15.925) | 0.115 | |
| T/S ratio + Years from Diagnosis + LTOT + DLCO | 0.170 | (0.036, 0.807) | 0.026 | |
| T/S ratio + Years from Diagnosis + LTOT + DLCO + FVC | 0.174 | (0.036, 0.846) | 0.030 | |
| T/S ratio + Years from Diagnosis + LTOT + DLCO + FVC (IPF patients only) | 0.096 | (0.011, 0.849) | 0.035 | |
Abbreviations: IPF: Idiopathic Pulmonary Fibrosis, IPF-AE: Idiopathic Pulmonary fibrosis Acute Exacerbation, CPFE: Combined Pulmonary Fibrosis and Emphysema, RA-UIP-ILD: Rheumatoid Arthritis-usual interstitial pneumonia associated Interstitial Lung Disease, UIP: Usual Interstitial Pneumonia, FVC: Forced Vital Capacity, DLCO: Diffusing Capacity of the lung for carbon monoxide, LTOT: long-term oxygen therapy, CRP: C-reactive protein, pCT: procalcitonin. Bold represent statistically significant differences at p < 0.05.
Association of T/S ratio with the appearance of IPF-AE.
| Binary logistic regression | Models | OR | 95% C.I. | p-value |
|---|---|---|---|---|
| Univariate Analysis | ||||
| T/S ratio groupsa | 8.500 | (2.060, 35.080) | 0.003 | |
| Age | 1.022 | (0.933, 1.120) | 0.636 | |
| Sex (Indicator: Male) | 1.750 | (0.432, 7.084) | 0.433 | |
| Years from Diagnosis | 1.577 | (1.081, 2.302) | 0.018 | |
| Smoking (Indicator:No) | 0.499 | |||
| Ex-Smoker | 1.429 | (0.388, 5.264) | 0.592 | |
| Current Smoker | 0.357 | (0.033, 3.916) | 0.399 | |
| CRP | 1.056 | (0.990, 1.126) | 0.097 | |
| pCT | 463.385 | (0.000, 3680.352) | 0.465 | |
| DLCO | 0.937 | (0.886, 0.991) | 0.023 | |
| FVC | 0.936 | (0.895, 0.979) | 0.004 | |
| Multivariate Analysis | ||||
| T/S ratio groups + Years from Diagnosis + DLCO + FVC | 30.787 | (2.153, 440.183) | 0.012 | |
Abbreviations: FVC: Forced Vital Capacity, DLCO: Diffusing Capacity of the lung for carbon monoxide, LTOT: long-term oxygen therapy, CRP: C-reactive protein, pCT: procalcitonin. Bold represent statistically significant differences at p < 0.05.
In this study shorter telomere length was detected in the peripheral blood of patients with sporadic IPF, IPF-AE, CPFE or RA-UIP-ILD, all associated with evidence of UIP pattern on HRCT compared to age-matched healthy controls. In addition, a significant association of TL with IPF-AE was found which was independent of age, gender, years from first diagnosis, smoking habit and lung function impairment. Moreover, in all UIP-fILD patients shorter TL was associated with increased risk of death. For IPF patients it was associated with reduced overall survival, an effect that was most consistently observed in IPF-AE.
Our results are consistent with previous studies, which have reported shorter telomeres in patients with IIPs,6 sporadic IPF,6–8,13–20 ILDs10,20 and RA-ILD.21,23 Interestingly, in line with the results of Stuart et al., no effect of TL on the survival of CPFE and RA-ILD associated with UIP pattern was observed in our study.13,14 So far it is well known that short telomeres concern not only sporadic IPF in which approximately 25% of patients have TL below the 10th percentile,13,16 but also may characterize other fibrotic subgroups of ILDs, such as CPFE and RA-UIP-ILD. However, to the best of our knowledge, no data exist concerning the association of TL with the UIP pattern. The association of short TL with the UIP pattern independently of the disease provenience found in the present study further reinforces the fact that the presence and extent of the UIP pattern is related to a worse prognosis.31
In the present study no difference was observed in TL among the diverse groups with a UIP pattern we examined, CPFE and RA-ILD included, compared to patients with stable IPF. This is apparently in contrast with the results of the study of Snetselaar et al.10 who demonstrated that patients with sporadic IPF had significantly shorter TL compared to those with idiopathic NSIP, smoking-related-ILD and CTD-ILD. However, the authors had not focused exclusively on the UIP pattern. CTD-ILD, especially, represents a heterogeneous disease, which may include diverse patterns of lung involvement, such as organizing pneumonia, desquamative interstitial pneumonia, fibrotic non-specific interstitial pneumonia, as well as UIP, thus, making it difficult to draw safe conclusions for the whole group independently of the prevailed radiological pattern.22,32,33 It is well known that in RA-ILD the UIP pattern predominates32 and in our study all RA-ILD patients were included based on the presence of UIP pattern. Our results, which did not demonstrate significant difference in TL between IPF and RA-UIP-ILD, are also supported by the fact that both diseases share common phenotypic similarities and are associated with poor prognosis.21,23,32,33 Furthermore, it was recently shown that both diseases share common genetic variant associations for the development of pulmonary fibrosis, including the gain-of-function MUC5B promoter variant.34
It is well known that chronic hypersensitivity pneumonitis may be confused with IPF.
As Nogueira and co-authors point out in their review,28 the identification of potential antigen sources the patient has been exposed to is essential for the diagnosis of HP. In addition, the authors state that the identification of antigen sources “…is often hampered by the lack of recognition of some sources and/or by the absence of standardized detection methods of sensitization.”28 In our study, to increase diagnostic accuracy and minimize misclassification we decided to use the questionnaire developed by the American College of Chest Physicians (CHEST)29 although it does not represent a standardized method, to uncover and exclude potential chronic HP cases.
Short telomeres are known to express the limited tissue renewal capacity in the lung.4,9,35 In IPF, a chronic lung disease characterized by irreversible fibrosis, repetitive causative stimuli force the bronchoalveolar epithelium to be constantly replaced.6,36 Thus, it is not surprising that short TL is found in IPF patients.6,13–20 Our results are compatible with those of previous studies, showing that IPF patients with no family history have significantly shorter TL compared to age-matched controls.6,13,15,16,18,19 Alder and co-authors showed that although mutations in the essential components of telomerase were detected in only 1% of individuals with sporadic form of IPF, TL represented an important risk factor for the development of the disease.6 Also, they found that IIPs patients had shorter telomeres both in peripheral blood and in the lung compared to age-matched controls.6 It remains uncertain whether CPFE represents a distinct entity, or is a coincidental presence of two smoking-related conditions, such as emphysema and fibrosis.37 Our results may provide some evidence that telomeropathy constitutes a potential explanation of its pathogenesis at least when it is associated with a UIP pattern on HRCT. However, further studies are needed to confirm whether telomere syndromes could be a risk factor for CPFE.
As mentioned above, several previous studies have investigated the pathogenetic and prognostic role of TL in the peripheral blood in stable IPF patients.13–17,19–21 However so far, to the best of our knowledge, there is a lack of studies on the role of TL in the appearance of IPF-AE, which represents the development of diffuse alveolar damage (DAD) upon UIP mostly triggered by microbes.24–26,38 The present study revealed that patients with IPF-AE had significantly shorter TL compared to the stable ones. Short telomeres have already been associated with DNA damage response resulting in apoptosis that clinically manifests as organ failure.9,33 In addition, it is well known that decreased TL can be the result either of the presence of increased oxidative stress or increased cell proliferation.39 A plausible explanation of the underlying pathogenetic mechanism for our finding concerning TL and IPF-AE may be that a potential decrease of TL below a critical threshold needed for lung repair is linked to the incapacity of lung tissue to regenerate after any trigger leading to this idiopathic deterioration and organ failure. Unfortunately, one of the limitations in this study is the limited number of patients, which did not allow us to examine whether TL shortens when IPF-AE occurs or is shorter since baseline. However, our observation that the increased risk of IPF-AE was independent of lung function impairment may denote that a short TL can be predictive of the risk of IPF-AE also at baseline even when the pulmonary function is not so compromised and the disease not so severe.
Another limitation of our study is that budget constraints prevented us from performing flow cytometry (flow-FISH). It should be noted that although this method has already been validated in previous studies, it is more expensive and technically demanding.40,41 On the other hand, real time quantitative PCR (qPCR) has been extensively adapted to measure TL; it is easier to perform, requiring only small amounts of DNA.42 However, our study represents a prospective, single-academic center study, based on a well-selected group of fibrotic ILD patients, which for the first time in Greek patients examined the potential association of TL with IPF-AE, CPFE and RA-UIP-ILD all associated with evidence of a UIP pattern on imaging.
ConclusionIn conclusion the present study shows that patients with sporadic ILD associated with a UIP pattern on imaging have shorter telomeres compared to age-matched controls. Furthermore, the study shows that patients experiencing IPF-AE present significantly shorter TL compared to the stable IPF ones, a relationship that is independent of age, gender, years from first diagnosis, smoking and lung function impairment. Also, in all UIP-f-ILDs patients shorter TL is associated with increased risk of death. Finally, as for IPF patients, shorter TL is associated with reduced overall survival and higher risk for IPF-AE or death. Large-scale studies are needed to confirm our results.
Author contributionsI.T., A.K. drafted the paper and were involved in study conception and design, data collection, data analyses, and data interpretation. E.D.M. was involved in data collection, data management and critical revision of the manuscript. C.K., A.S. and S.A. were involved in data analyses, data interpretation and critical revision of the manuscript. S.A.P. was involved in data interpretation and critical revision of the manuscript. All authors read and approved the final version of the submitted manuscript.
Funding sourcesThis work was supported by a Grant of the Hellenic Thoracic Society.
Conflicts of interestNo conflict of interest to declare for all authors.
This paper is dedicated to the memory of Professor P. Karakitsos, Director of the Department of Cytopathology, in “ATTIKON” University Hospital, at the National and Kapodistrian University of Athens, a great mentor and colleague who suddenly passed away during the study. The present study was funded by


