




Interstitial Lung Diseases (ILD) are a group of diseases characterized by bilateral involvement of lung parenchyma, acutely or chronically, which have different degrees of fibrosis and tissue inflammation and which occur in immunocompetent hosts, without infection or neoplasia.1
Bronchoalveolar lavage (BAL) is a useful diagnostic tool for ILD.2–4 The cellular profile obtained by BAL, in association with medical history, physical examination and imaging findings, may support or help narrowing the probable diagnosis hypothesis.1,3
We have conducted a retrospective observational study, in a pulmonology department of a university hospital, collecting demographic, clinical and laboratory data, consulting the clinical process of patients undergoing BAL on suspicion of ILD, during a period of 3 years and 7 months. The statistical analysis was performed using SPSS 20®.
The study included 188 patients, of whom, 125 had a definitive diagnosis of interstitial lung disease. The average age was 54.5 years old and 56% of patients were male.
In this study, it was our purpose to analyze both the alveolar cellular profile of the main interstitial lung diseases and the impact of the BAL in the initial diagnosis which was suggested by clinical features and high resolution computed tomography (HRCT).
Cellular analysis of BAL revealed BAL neutrophilia in patients with connective tissue associated ILD (ILD-CTD) and in patients with idiopathic pulmonary fibrosis (IPF) with a differential cellular count of 21% and 20%, respectively (Fig. 1). Neutrophilia and eosinophilia are more common in BAL of ILD that are characterized by fibrosis (as a whole, Idiopathic interstitial fibrosis), which match the results obtained.5,6 Significant lymphocytosis was observed in BAL of patients with sarcoidosis, hypersensitivity pneumonitis, cryptogenic organizational pneumonitis, pneumoconiosis and non-specific interstitial pneumonia, who presented differential cellular count of 48.0%, 47.7%, 38.6%, 37.3% and 33.5%, respectively (Fig. 1).
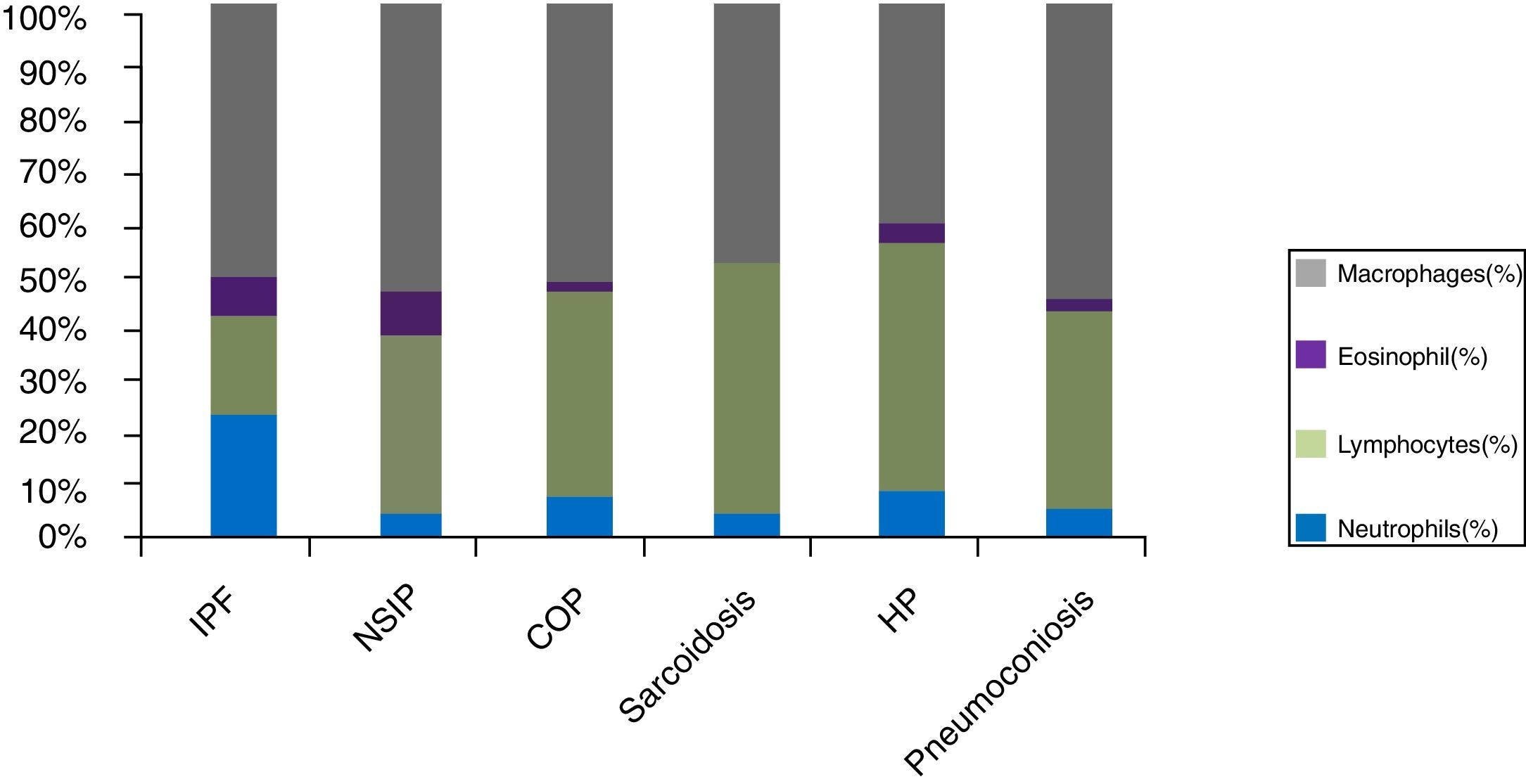
ILDs characterized by a granulomatous pattern, such as hypersensitivity pneumonitis and sarcoidosis, are characterized by an increase in lymphocytes number, with variable neutrophilia and occasionally eosinophilia (Fig. 1).5
However, as observed in the obtained results, idiopathic interstitial pneumonias such as cryptogenic organizing pneumonia (COP) and nonspecific interstitial pneumonia (NSIP) also have lymphocytosis in BAL (Fig. 1).1,2,4 In our study, patients with NSIP had mostly a cellular-form of the disease which may explain the BAL lymphocytosis profile in this group.
Lymphocytosis superior to 15% in BAL diminishes the probability of idiopathic pulmonary fibrosis (IPF) and may shift differential diagnosis towards other entities: NSIP, COP or chronic HP.6,7
In COP, usually, there is an increase in the number of lymphocytes (up to 40%), an increase of neutrophils (mainly in early stages of disease) and eosinophils (Fig. 1).7
It was established that the contribution of BAL to the final diagnosis could present, for each patient, one of the following possible results:
Confirm the initial diagnosis - the BAL result made the definitive diagnosis or was highly suggestive of the initial diagnosis, with no further examinations needed;
Support the initial diagnosis - the BAL result was consistent with the initial diagnosis, but other procedures were required to confirm it;
Suggest another diagnosis - the BAL result suggested another diagnosis, leading the investigation into another way;
Exclude the initial diagnosis - the BAL result was inconsistent with the initial diagnosis hypothesis or established, definitively, another diagnosis;
No contribution - the BAL result had no diagnostic value to the investigation of the disease under study;
BAL confirmed the diagnosis, initially based on clinical and radiological data, in 60% of patients with pneumoconiosis, 45% of patients with hypersensitivity pneumonitis and 35% of patients with sarcoidosis. It also made it possible to exclude the diagnosis of IPF in 12% of patients. BAL did not contribute to the final diagnosis in 8% of the total patients (Fig. 2).
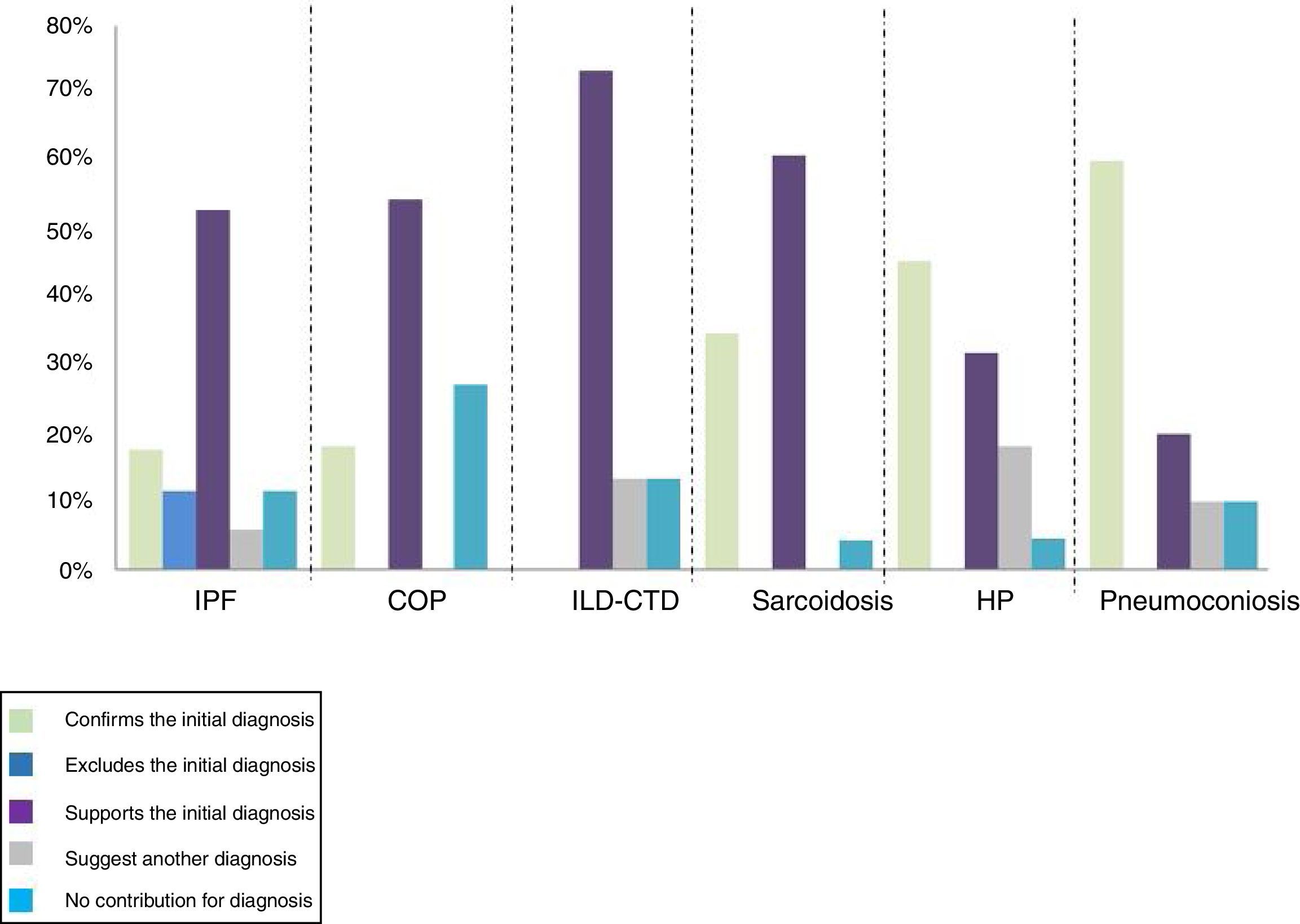
There were statistically significant differences in COP (z=-3.61, p<.001), in sarcoidosis (z=- 4.6, p<.001), in PH (z=5.01, p<.001), in CTD-ILD (z=-3.2, p=.001), in IPF (z=-3.4, p=.001) and in vasculitis (z=-2.2, p=.025), suggesting that, in these group of diseases, BAL revealed a greater contribution in the diagnosis.
BAL significant lymphocytosis (>40%) is expected in most patients with HP and, in appropriate clinical/radiological setting, may confirm the diagnosis. In our study, only in less than half of HP patients was BAL the determinant. This may be explained by a high proportion (>50%) of patients with chronic forms of the disease with extensive interstitial fibrosis and therefore with lower BAL lymphocyte count. In these patients differential diagnosis with other interstitial fibrotic diseases (like IPF) is problematic and multidisciplinary discussion is mandatory.
We conclude that BAL in our setting, played an important role in the diagnostic evaluation of different ILD, in carefully selected patients and under a multidisciplinary approach.
Conflict of interestsThe authors have no conflicts of interest to declare.


