






Idiopathic pulmonary fibrosis is a rare interstitial lung disease included in the Idiopathic Interstitial Pneumonias group. Although several potential risk factors have been described, it is a progressive fibrosing disease of unknown cause affecting mainly adults over 50 years and associated with a poor prognosis, reflected in a median survival of 2–3 years after diagnosis.
The concept of a multidisciplinary working group for the diagnosis of idiopathic pulmonary fibrosis is based on the need to have experienced pulmonologists, radiologists and pathologists in the evaluation and correct treatment of the disease, and requires the use of all available data about individual patients, standardized (largely through High Resolution Computed Tomography and pathology when needed) as well as non-standardized data (laboratory, serology and biomarkers). This approach helps to increase diagnostic accuracy and is an internationally accepted recommendation.
In regard to therapy, the situation has changed radically since the publication of the ATS/ERS/JRS/ALAT 2011 guidelines on the diagnosis and management of idiopathic pulmonary fibrosis where it was stressed that no proven therapy exists for this disease.
Currently besides non-pharmacological treatment, therapy of complications and comorbidities and palliative care, nintedanib and pirfenidone, two compounds with pleiotropic mechanisms of action, are to date, the two drugs with confirmed efficacy in slowing functional decline and disease progression in idiopathic pulmonary fibrosis patients.
Idiopathic pulmonary fibrosis (IPF) is the most common and fatal of the idiopathic interstitial pneumonias subgroup.1, 2, 3, 4, 5
After the first IPF consensus6 of the American Thoracic Society (ATS)/European Respiratory Society (ERS) in 2000, several studies contributed to a greater knowledge of IPF diagnosis and treatment, which was reflected in the updated international consensus on the diagnosis and management of IPF published in 2011 and signed by the American (ATS), European (ERS), Japanese (JRS) and Latin American Associations (ALAT).1 Since this publication several developments in the IPF management there have been, including diagnostic criteria and therapeutic options.7, 8, 9, 10, 11
Interstitial lung diseases are identified as priority intervention in the Portuguese National Program of Respiratory Diseases12; nonetheless there is a general consensus about the need for greater awareness for the identification of these patients, enabling an earlier diagnosis and treatment.
DefinitionIPF is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause, predominantly in adults over 50 years, limited to the lungs, and associated with the radiologic and/or histopathologic pattern of usual interstitial pneumonia (UIP).1, 2, 3, 13
EpidemiologyDepending on the criteria and methodology adopted in reliable studies,14 IPF incidence ranges between 0.22 and 17.4 per 100,000 person-year, and the prevalence between 14 and 27.9 cases per 100,000 persons. It is unknown if geographic, ethnic, cultural or racial factors influence the incidence and prevalence of IPF.1, 15 IPF is associated with a poor prognosis, with a median survival of 2–3 years after the diagnosis.16, 17, 18
Risk factorsAlthough the aetiology of IPF is unknown, several potential risk factors have been identified including: (a) environmental factors like smoking history of >20 packs/year, exposure to silicon, brass, steel, lead and wood dust, farming and agricultural work, construction of wooden houses19; (b) genetic factors such as familial pulmonary fibrosis, mutations in the genes that maintain the length of the telomeres (TERT – Telomerase reverse transcriptase, TERC – Telomerase RNA component), mutations in the surfactant protein C gene and mutations in the mucin 5B promoter region (MUC5B)20, 21, 22, 23; (c) gastro-oesophageal reflux (GER), considered a risk factor for the predisposition and progression of IPF24, 25; (d) viral infections, although there is no sufficient data to state their role in IPF despite the large number of studies analyzing the etiological risk effect of hepatitis C virus, herpes virus and adenovirus26, 27; and (e) autoimmunity, that is still being studied but the possible autoimmune origin of IPF is based on the association of radiologic and/or histologic manifestations of UIP to connective tissue diseases.1, 13
DiagnosisThe diagnosis of IPF requires a multidisciplinary approach, including the awareness of general practice physicians, and experienced pulmonologists, radiologists and pathologists in the evaluation and correct treatment of diffuse interstitial lung disease (DILD).1, 3, 13, 28, 29, 30, 31 This approach helps increase diagnostic accuracy and is an internationally accepted recommendation.28, 29
Table 1 presents the procedures that should be performed at diagnosis, systematically and occasionally.
Table 1. Idiopathic pulmonary fibrosis diagnosis examinations.
| At IPF diagnosis |
| Systematic |
| Chest HRCT |
| Plethysmography and DLCO |
| Arterial blood gas in room air at rest |
| 6MWT with measurement of percutaneous oxygen saturation |
| Doppler echocardiography |
| Biology |
| Antinuclear antibodies |
| Anti-citrullinated cyclic peptides antibodies |
| Rheumatoid factor |
| Extractable Nuclear Antigens antibodies |
| Occasional |
| Video-assisted surgical lung biopsy |
| Differential cell count of BAL |
| Depending on context |
| Genetic testing |
| Anti-neutrophil cytoplasmic antibodies |
| Creatine phosphokinase |
| Anti-thyroid antibodies |
| Precipitins (depending on disease presentation) |
| Electrophoresis of serum proteins, immuno-electrophoresis of serum proteins, urine immunofixation and cryoglobulinaemia |
| Exploration for gastro-oesophageal reflux |
| Exploration for sleep apnoea syndrome |
Adapted from Cottin et al. 3
6MWT: 6-min walk test; BAL: bronchoalveolar lavage; DLCO: diffusing capacity of the lung for carbon monoxide; HRCT: high-resolution computed tomography.
The general practice physician must be aware of IPF symptoms in order to consider this disease when establishing the initial diagnosis, and to refer IPF suspicious cases to a pulmonologist.
The diagnosis of IPF requires the exclusion of other known causes of interstitial lung disease (domestic or occupational environmental exposures, connective tissue disease, drug toxicity) and the presence of a typical histological UIP pattern.1, 3, 7, 13, 18, 32 According to the ATS/ERS recommendations and validated by international societies, in the absence of biopsy, IPF can be diagnosed only based on radiological criteria from the high resolution computed tomography (HRCT).1
Figure 1 presents the algorithm proposed for the diagnosis of IPF. In case of IPF suspicion, all the possible associations with UIP radiological pattern must be accurately investigated. Since UIP pattern can be associated with several autoimmune diseases, such as rheumatoid arthritis, systemic sclerosis or Sjögren syndrome or even, although rarely, with ANCA-positive vasculitis, all patients should perform serological autoimmune tests, although positive anti-nuclear antibodies or rheumatoid factor can be detected in up to 20% of IPF cases as an unspecific feature with no association with any particular disorder.1, 32, 33, 34 The presence of serum specific IgG against the antigens that can frequently cause hypersensitivity pneumonitis should also be systematically assessed.1, 30, 34, 35
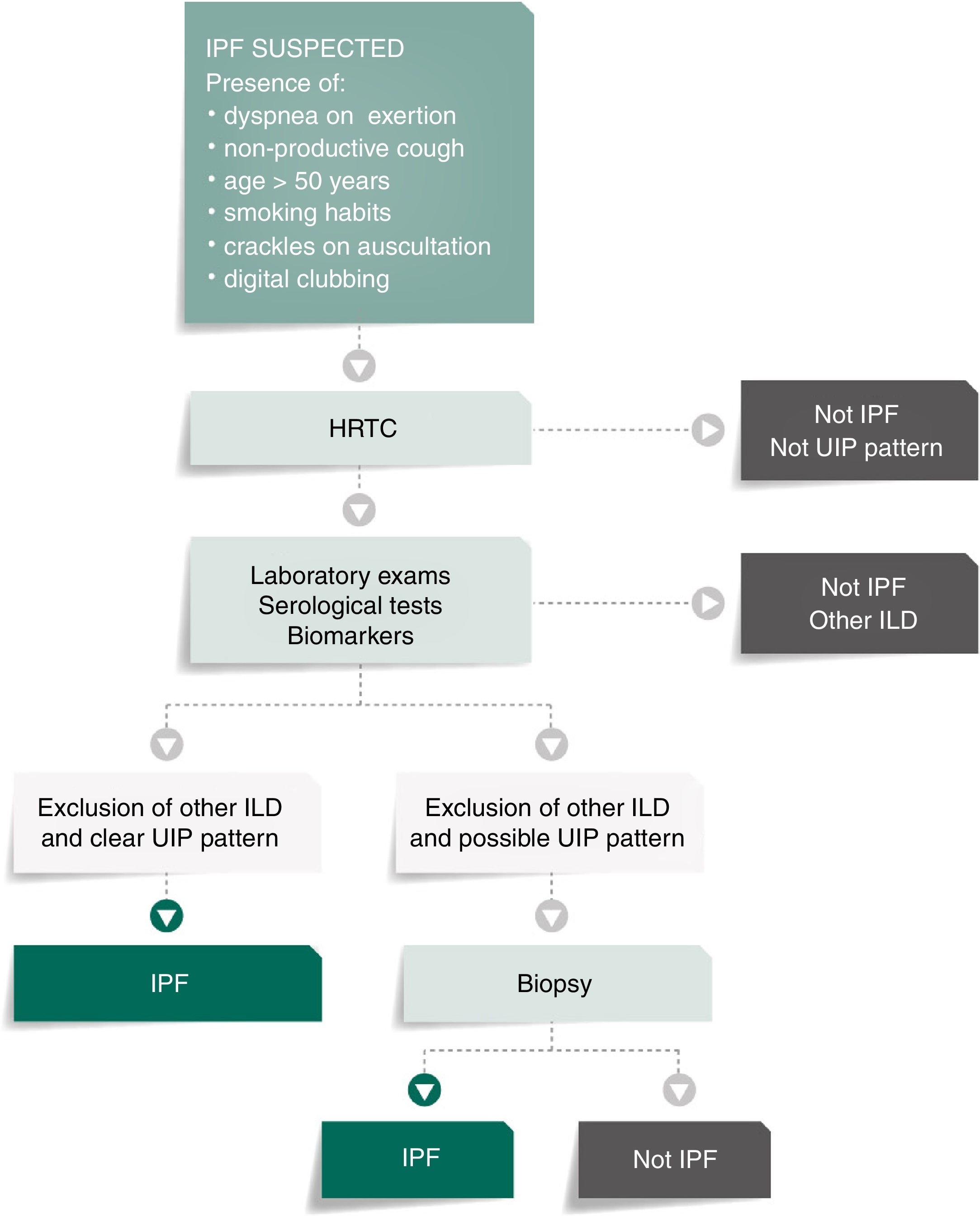
Figure 1. IPF diagnosis algorithm. ILD: interstitial lung disease; IPF: idiopathic pulmonary fibrosis; HRCT: high-resolution computed tomography; UIP: usual interstitial pneumonia.
Bronchoalveolar lavage (BAL) is not included in this algorithm as a diagnostic procedure since it is not recommended that BAL be systematically performed in all patients, although it may be appropriate for a minority of cases.3, 36, 37, 38, 39 Nevertheless, Centres with experience in this approach might use BAL regularly as it can either rule out or help diagnose IPF, especially when a higher lymphocytosis guides the diagnosis for other entities, as for instance hypersensitivity pneumonitis.37 On the other hand, the use of transbronchial biopsy in the diagnosis of IPF is controversial. Cryobiopsy, although it is not yet widely used and must only be performed in Centres with experience in this procedure, the published data so far shows that cryobiopsy in fibrotic ILDs yields large and well-preserved lung specimens, sufficient to enclose all the UIP characteristic features, with safety and tolerability.40, 41, 42 In a recent cross-sectional study, a comparison of cryobiopsies with surgical lung biopsies in fibrotic ILD by a multidisciplinary committee, conclude a major increase in diagnostic confidence after the addition of cryobiopsies similar to surgical lung biopsy (SLB) in IPF diagnosis (from 29% to 63%, p = 0.0003 and from 30% to 65%, p = 0.0016 of high confidence in the bronchoscopic lung cryobiopsy group and SLB group respectively), which constitutes robust evidence of the potential role of this bronchologic procedure.43
High resolution computed tomographyThe purpose of HRCT is to identify the typical pattern of changes of UIP (Table 2) or other patterns that may be present, supporting the diagnosis of other lung diseases. To standardize these radiological criteria, the report should use descriptive concepts based on the terminology recommended by Fleischner Society.44
Table 2. High-resolution computed tomography's idiopathic pulmonary fibrosis/usual interstitial pneumonia diagnosis criteria.
| UIP pattern (all of the following) | Possible UIP pattern (all of the following) | UIP-IPF pattern (1 + 2) |
| 1-Subpleural, basal (predominant distribution) | 1-Subpleural, basal (predominant distribution) | 1-All of UIP pattern |
| 2-Reticulation | 2-Reticulation | 2-Absence of findings that do not support a UIP pattern |
| 3-Honeycombing with or without traction bronchiectasis | 3-Absence of findings that do not support a UIP pattern |
Adapted from Raghu et al. 1
IPF: idiopathic pulmonary fibrosis; UIP: usual interstitial pneumonia.
The identification of lung honeycombing is crucial for the diagnosis of IPF – they are cystic axial, confluent or layered images, with diameters of 3–10 mm, reaching 2.5 cm. The distribution is usually subpleural or in the reticulation areas.45, 46, 47, 48, 49
There are two aspects that may raise doubts in the HRTC interpretation: the distinction between honeycombing and traction bronchiolectasis and fibrosis with emphysema. The differential diagnosis is a challenge even for an experienced radiologist.47, 49, 50 The 2011 ATS/ERS/JRS/ALAT multidisciplinary consensus1 stated that the identification of four typical findings establishes the definite diagnosis of UIP: 1 – subpleural and basal lung lesions predominance; 2 – reticulation; 3 – honeycombing with/without traction bronchiectasis/bronchiolectasis; 4 – absence of findings that do not support a UIP pattern (Table 2, Table 3; Figure 2).
Table 3. Inconsistent criteria in the high-resolution computed tomography's idiopathic pulmonary fibrosis/usual interstitial pneumonia diagnosis.
| Any of the following |
| Predominance of middle or upper distribution |
| Peri-bronchovascular distribution |
| Reticular ground glass opacities |
| Micronodules (bilateral and upper lobes predominance) |
| Axial cysts (multiple, bilateral, distant from the areas of honeycombing) |
| Mosaic pattern, diffuse/air trapping (bilateral, in three or more lobes) |
| Segment/lobar consolidation |
Adapted from Raghu et al. 1
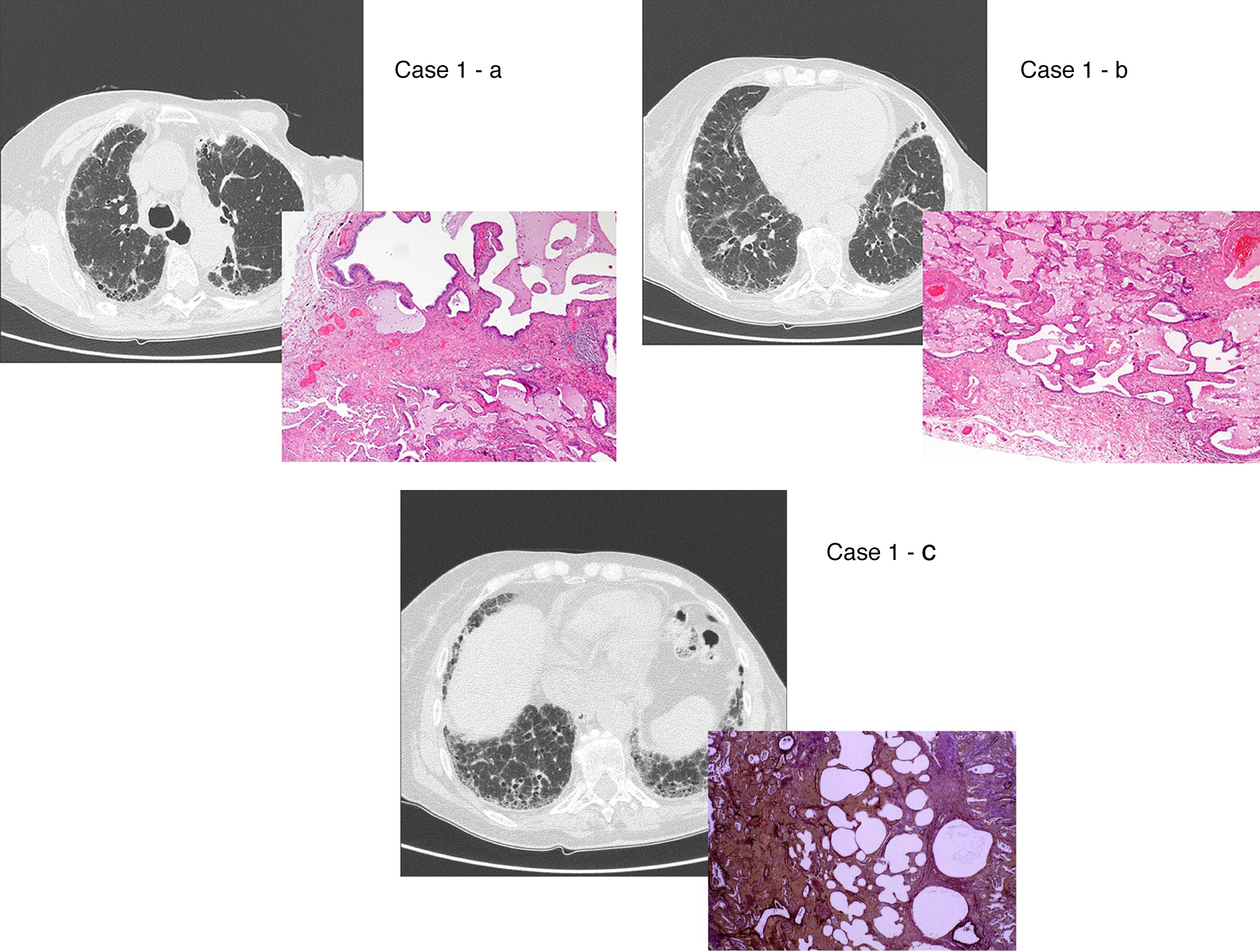
Figure 2. UIP/IPF identification in HRCT and histopathology. Case 1 – a: Common histopathological features of UIP/IPF consisting in honeycombing as a result of subpleural airway spaces confluence with bronchialization of epithelium where mucus cells may be predominant. HE 100×. Case 1 – b: The previous image aspects are revealed by higher production of mucus occupying the new formed subpleural smaller confluent airspaces. HE 100×. Case 1 – c: The interstitium intermingling honey-combing loses elastin fibres and is represented by fusiform cells, either fibroblasts or miofibroblasts, with collagen deposition. Elastin-van Gieson 100×. Case 2 – a: Irregular confluent air spaces with typical subpleural localization of UIP/IPF honeycombing with interstitium enlargement by fusiform cells. HE 100×. Case 2 – b: The adjacent lobular parenchyma accentuate histopathological heterogeneity, starting by overinflation and pseudo emphysema morphology. HE 100×. Case 2 – c: The lobular histopathological remodelling aspect of fibrosis enlarging alveoli septae till central bronchiolo-vascular axes and alveolar bronchialization. HE 100×. Case 3 – a: Small subpeural confluent air-spaces with bronchial-like epithelium and juvenile foci of fibroblasts in myxoid matrix; lymphocytes are seen. PAS 200×. Case 3 – b: UIP/IPF heterogeneous morphology in airspaces confluence with subpleural preponderance and committing the whole lobule. HE 100×.
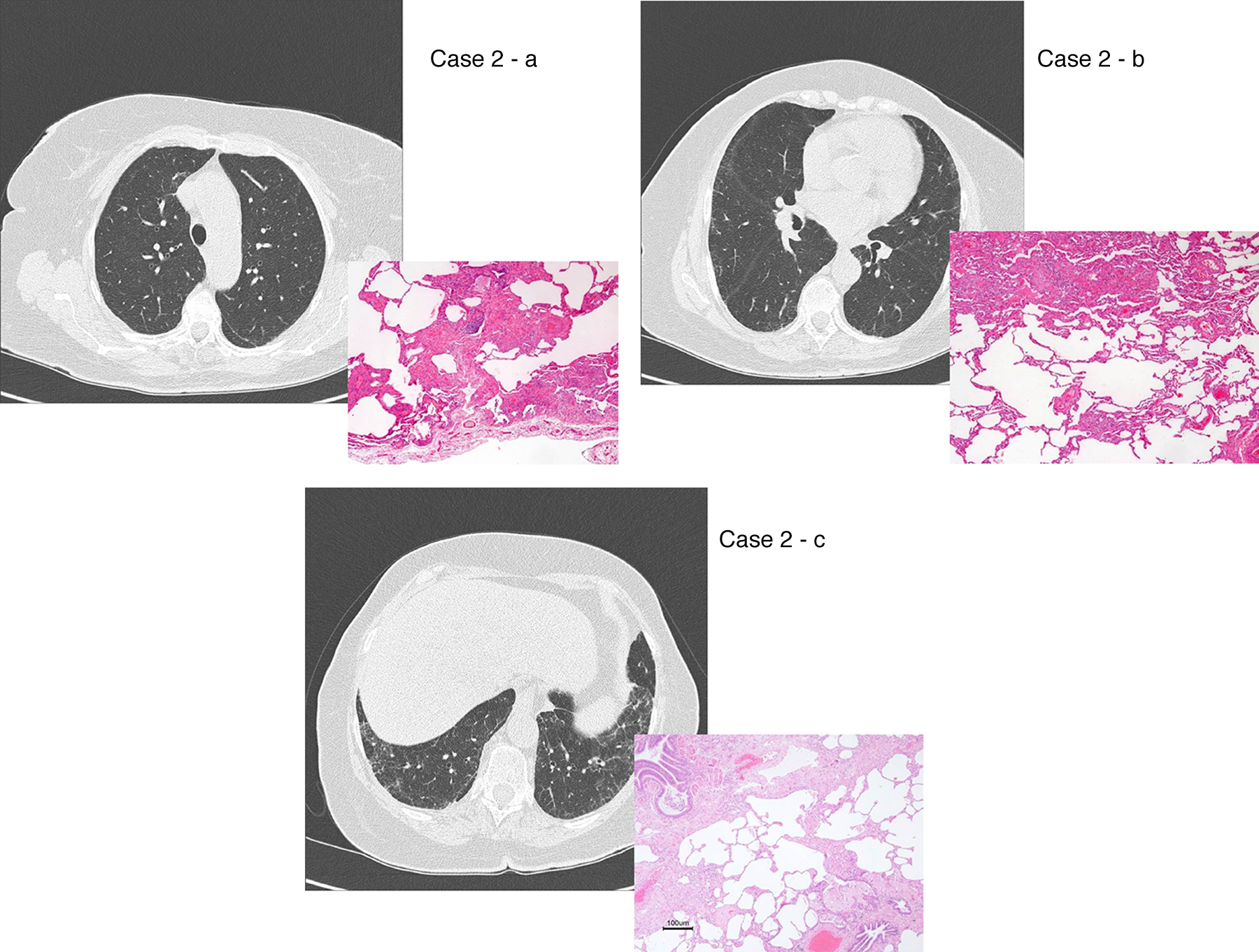
Figure 2. UIP/IPF identification in HRCT and histopathology. Case 1 – a: Common histopathological features of UIP/IPF consisting in honeycombing as a result of subpleural airway spaces confluence with bronchialization of epithelium where mucus cells may be predominant. HE 100×. Case 1 – b: The previous image aspects are revealed by higher production of mucus occupying the new formed subpleural smaller confluent airspaces. HE 100×. Case 1 – c: The interstitium intermingling honey-combing loses elastin fibres and is represented by fusiform cells, either fibroblasts or miofibroblasts, with collagen deposition. Elastin-van Gieson 100×. Case 2 – a: Irregular confluent air spaces with typical subpleural localization of UIP/IPF honeycombing with interstitium enlargement by fusiform cells. HE 100×. Case 2 – b: The adjacent lobular parenchyma accentuate histopathological heterogeneity, starting by overinflation and pseudo emphysema morphology. HE 100×. Case 2 – c: The lobular histopathological remodelling aspect of fibrosis enlarging alveoli septae till central bronchiolo-vascular axes and alveolar bronchialization. HE 100×. Case 3 – a: Small subpeural confluent air-spaces with bronchial-like epithelium and juvenile foci of fibroblasts in myxoid matrix; lymphocytes are seen. PAS 200×. Case 3 – b: UIP/IPF heterogeneous morphology in airspaces confluence with subpleural preponderance and committing the whole lobule. HE 100×.
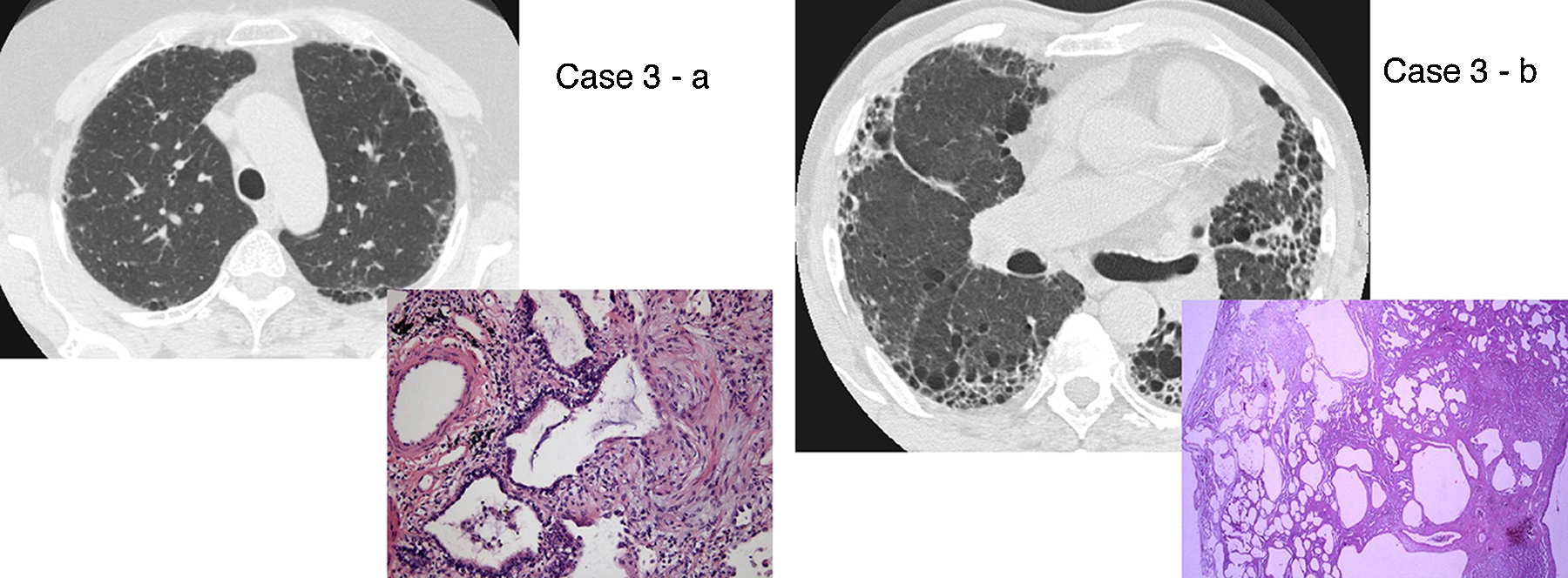
Figure 2. UIP/IPF identification in HRCT and histopathology. Case 1 – a: Common histopathological features of UIP/IPF consisting in honeycombing as a result of subpleural airway spaces confluence with bronchialization of epithelium where mucus cells may be predominant. HE 100×. Case 1 – b: The previous image aspects are revealed by higher production of mucus occupying the new formed subpleural smaller confluent airspaces. HE 100×. Case 1 – c: The interstitium intermingling honey-combing loses elastin fibres and is represented by fusiform cells, either fibroblasts or miofibroblasts, with collagen deposition. Elastin-van Gieson 100×. Case 2 – a: Irregular confluent air spaces with typical subpleural localization of UIP/IPF honeycombing with interstitium enlargement by fusiform cells. HE 100×. Case 2 – b: The adjacent lobular parenchyma accentuate histopathological heterogeneity, starting by overinflation and pseudo emphysema morphology. HE 100×. Case 2 – c: The lobular histopathological remodelling aspect of fibrosis enlarging alveoli septae till central bronchiolo-vascular axes and alveolar bronchialization. HE 100×. Case 3 – a: Small subpeural confluent air-spaces with bronchial-like epithelium and juvenile foci of fibroblasts in myxoid matrix; lymphocytes are seen. PAS 200×. Case 3 – b: UIP/IPF heterogeneous morphology in airspaces confluence with subpleural preponderance and committing the whole lobule. HE 100×.
The HRCT positive predictive value in the diagnosis of UIP is 90–100%. The histopathology may also establish a UIP diagnosis in patients with no UIP lesions in the HRTC.
The 2011 official ATS/ERS/JRS/ALAT consensus established the use of “definitive” and “possible” IPF; these definitions simplify the definitive diagnosis except for the other patients. According with to this multidisciplinary consensus, the diagnosis of UIP/IPF must be based on the consensus between the clinician, the radiologist and the pathologist.1
HistopathologyA surgical lung biopsy should establish the diagnosis of IPF if there are doubts about the presence of a definitive UIP typical pattern in the HRCT interpretation. In patients with suspected IPF, surgical lung biopsies should be obtained from multiple lobes, avoiding the extremity of the lingula and middle lobe.1, 51
The histological pattern of UIP must present: 1 – marked fibrosis/architectural distortion, with or without honeycombing in a predominantly subpleural/paraseptal distribution; 2 – patchy involvement of lung parenchyma by fibrosis; 3 – fibroblast foci; 4 – absence of features against a diagnosis of UIP suggesting an alternate diagnosis1, 51 (Figure 2).
In some diseases, like rheumatoid arthritis, chronic hypersensitivity pneumonitis or drug-induced pneumonitis, asbestosis and familial fibrosis, a histological pattern indistinguishable from UIP can be observed, so the presence of granulomas, asbestos bodies, specific infections or other exogenous agents must be discarded in the biopsy.13, 51
The biopsy should only be performed after a multidisciplinary meeting where IPF diagnosis was not defined by HRCT; if the biopsy is performed, its results should again be discussed in a multidisciplinary meeting.51
Follow-upIt is recommended that IPF patients perform a follow-up clinic visit every 3–6 months. Table 4 presents the main examinations that are useful for IPF management.
Table 4. Idiopathic pulmonary fibrosis management examinations.
| During IPF follow-up |
| Every 6 months |
| FVC and DLCO |
| Arterial blood gas in room air |
| 6MWT |
| Chest X ray |
| Depending on context |
| Chest HRCT |
| Doppler echocardiography |
| TLC |
| Right heart catheterization |
Adapted from Cottin et al. 3
6MWT: 6-min walk test; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity; HRCT: high-resolution computed tomography; TLC: total lung capacity.
Setting an IPF patient flow, which defines the several steps from diagnosis to treatment, may represent the identification of more cases of IPF, in a shorter period of time and in early stages of the disease (Figure 3).

Figure 3. Idiopathic pulmonary fibrosis patient flow. HRCT: high-resolution computed tomography.
Pharmacological treatmentThe therapeutic strategy in IPF should be individualized for each patient, considering the evaluation of potential benefits and risks. It is more important to take into account the clinical behaviour than the respiratory function parameters.52
Nintedanib and pirfenidone, two compounds with pleiotropic mechanisms of action are, to date, the two drugs with confirmed efficacy in slowing functional decline and disease progression in IPF patients.7, 53, 54
NintedanibNintedanib is a tyrosine kinase inhibitor small molecule that targets receptors of growth factors, specifically inhibits platelet-derived growth factor receptor (PDGFR), fibroblasts growth factor receptor (FGFR) and vascular endothelial growth factor receptor (VEGFR),55, 56, 57 involved in the lung fibrosis mechanism. It is believed that nintedanib reduces the progression of the disease and the decline in lung function by blocking the signalling pathways involved in the fibrotic processes.55, 56, 57
The TOMORROW Phase II trial showed that nintedanib, 150 mg twice daily, was effective in reducing forced vital capacity (FVC) decline, in preventing acute exacerbations, and preserving health-related quality of life (HRQoL).9 The phase III INPULSIS-1 and INPULSIS-2 trials, showed that nintedanib slowed disease progression, reducing the annual rate of lung function decline of 50%.11 It also presented a 68% reduction of adjudicated acute exacerbations,11 which can be crucial since approximately 50% of IPF acute exacerbation admitted patients die during hospitalization.58 After the first publication further subgroup analysis of pooled data from the INPULSIS trials demonstrated that nintedanib slowed disease progression by reducing the annual rate of FVC decline independently of the FVC value (FVC >80% versus <80% or FVC >70% versus <70%), of the presence of emphysema at baseline, of the presence of honeycombing and/or biopsy confirmation of UIP or in the absence of both, namely the presence of “possible UIP” on HRCT scan without histology confirmation.
Although these studies were not dimensioned to detect statistical differences in mortality, the pooled analysis of data from the TOMORROW and INPULSIS trials shows a trend towards reduction in mortality, since after 52 weeks the proportion of patients who died from any cause was 5.8% in the nintendanib arm and 8.3% in the placebo group (HR 0.70 [95% CI: 0.46, 1.08]; p = 0.0955). Respiratory mortality was also comparatively low in the nintendanib arm (3.6% versus 5.7% – HR 0.62 [95% CI: 0.37, 1.06]; p = 0.0779).11, 59
The most frequent adverse events during these trials were diarrhoea, nausea and vomiting, that can be easily managed in most patients.11
Nintedanib is approved in the US and EU for the treatment of IPF patients, regardless of disease severity, under the brand name OFEV®.
PirfenidonePirfenidone is a pyridine compound with antifibrotic and anti-inflammatory properties, indicated for the treatment of mild to moderate IPF.60 The mechanism of action of pirfenidone has not been fully established, but it is known that pirfenidone attenuates fibroblast proliferation, production of fibrosis-associated proteins and cytokines, and the increased biosynthesis and accumulation of extracellular matrix in response to cytokine growth factors such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF).60
Despite the conflicting outcomes related to FVC decline in the CAPACITY trials 004 and 006, the pooled data 8 showed a pirfenidone treatment effect in percentage predicted FVC at week 72 with a dose of 2403 mg/day (−8.5% versus −11.0%; p = 0.005) and a smaller proportion of patients with a FVC decline ≥10%. Pirfenidone also prolonged progression free survival by 23% when compared with placebo.8 A 31% relative difference favouring pirfenidone8 was noted related to 6-min walk test (6MWT) also at week 72. The posterior phase III ASCEND trial showed a reduction of 47.9% of patients with a FVC percentage predicted value decline ≥10% or who died.10 There was also an increase of 132.5% in proportion of patients with no decline in FVC (p < 0.001).10 A reduced decline in 6MWT (p = 0.04) and an improved progression free survival (p < 0.001) were also noted.10
Moreover, in a pooled analysis enclosing CAPACITY and ASCEND trials, there was a significant difference in death cases from any causes between patients treated with pirfenidone and placebo (p = 0.01) and death related with IPF (p = 0.006).10
In a recent pooled data analysis of outcomes at 1 year in CAPACITY and ASCEND trials including a total of 1247 patients pirfenidone reduced the proportion of patients with a ≥10% decline in FVC percentage or death by 43.8% (95% CI 29.3–55.4%) and increased the proportion of patients with no decline by 59.3% (95% CI 29.0–96.8%). Progression free survival, 6MWT and dyspnoea also showed a positive impact from pirfenidone. These results provided further evidence about the benefit of pirfenidone in IPF disease progression.61
The most common adverse events during these trials were photosensibility, gastro-intestinal occurrences and gastro-oesophageal reflux, generally reversible with dose reductions.8, 10, 62, 63, 64
Pirfenidone is approved for the treatment of mild to moderate IPF patients, under the brand name of Esbriet®.
Although some data is available related with the concomitant therapy with nintedanib and pirfenidone, this approach cannot be recommended since its efficacy and tolerability is not sufficiently evidenced.65
Other drugsAfter the IFIGENIA study,66 N-acetylcysteine (NAC) combined with prednisone and azathioprine was the treatment of choice for several years.1 With the presentation of the subsequent PANTHER study,67 which showed higher mortality and hospital admissions in patients receiving NAC combination therapy compared to placebo or NAC alone, the triple therapy is not recommended.1 Additionally, NAC as monotherapy did not show efficacy versus placebo.68
Various compounds, including anti-coagulants, interferon gamma-1b, anti-acids, anti-inflammatory and vasodilator agents25, 40, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 have been tested in IPF patients but have not demonstrated definitive efficacy for the treatment of IPF. In addition, there are currently several ongoing clinical trials results of which will be available in the near future.
Non-pharmacological treatmentRespiratory rehabilitationPulmonary rehabilitation in IPF has showed an improvement in walking distance, symptoms and HRQoL. Although the long term benefit of rehabilitation is not demonstrated, it is recommended for the majority of patients, namely in early stages of the disease.1, 82, 83
Oxygen therapyAlthough there are no specific data concerning the use of long-term oxygen therapy in patients with IPF, based on chronic obstructive pulmonary disease (COPD) and chronic respiratory failures studies, it is recommended to all IPF patients with resting hypoxaemia.1
Lung transplantIn cases that meet the criteria, lung transplant must be always kept in mind since it is, according to the international recommendations, the most effective and reliable treatment for patients with IPF. Lung transplantation 5-years survival rate ranges from 50% to 56% and 10-years survival rate is 30%.1, 84, 85
Treatment of complications and comorbiditiesAcute exacerbationHigh-dose corticosteroid therapy with or without immunosuppressive agents, namely cyclophosphamide, and wide spectrum antibiotics, are the most widely used treatments for the acute exacerbation of IPF despite the lack of conclusive evidence demonstrating their benefits.1, 86, 87
Gastro-oesophageal refluxAsymptomatic GER is common in IPF and may impact IPF natural history and progression.88 An analysis of data from three randomized controlled trials showed that IPF patients taking proton pump inhibitors (PPI) and/or H2 inhibitors significantly slower decline in FVC over time.25 According to the international recommendations, asymptomatic GER should be treated in the majority of patients with IPF, namely with PPI.1, 53
Pulmonary hypertensionIn two uncontrolled prospective studies of sildenafil in patients with pulmonary hypertension in context of IPF, walk distance and pulmonary haemodynamics improved after 8–12 weeks of treatment.89, 90 In the randomized controlled STEP-IPF (Sildenafil Trial of Exercise Performance in IPF) trial, comparing 20 mg tid of sildenafil and placebo over 12 weeks, no significant improvement of the 6MWT (primary end-point) was observed; however arterial oxygenation, diffusing capacity of the lung for carbon monoxide (DLCO), dyspnoea and quality of life were improved in patients on sildenafil.80
However, in a subgroup analysis of patients with right ventricular systolic dysfunction echocardiogram-documented, sildenafil treatment resulted in a significant improvement in the primary outcome of 6MWT (mean distance, 99.3 m; 95% CI 22.3–176.2 m).91 Considering these results, although without a clear recommendation, sildenafil can be one option in patients with IPF and pulmonary hypertension with right ventricular dysfunction.
Due to lack of evidence, any drug from prostacycline analogues or endothelin receptor antagonists (ERA) are not indicated for use in IPF.52 Moreover, ambrisentan, an ERA, is contraindicated in IPF, since the ARTEMIS-IPF randomized controlled trial showed an increase number of hospitalizations for respiratory complications.92
Palliative careThe aim of palliative care is to improve patients’ quality of life. It is important to take into account not only the symptoms related with the disease progression, like cough and dyspnoea, where codeine and morphine have shown to be effective, but also psychological aspects inherent to those who face this poor prognostic and great disabling disease.1, 93, 94, 95
ConclusionsTo change the scenario of this devastating disease, early identification and referral of IPF patients to a specialized centre is needed. Training of General Practitioners and further awareness of pulmonologists not specialized in IPF is essential to achieve this.
Every patient older than 50 years of age, with exertional dyspnoea and dry, non-productive cough, with ongoing or previous smoking habits and bi-basal crackles on auscultation, should have rapid access to a multidisciplinary team.
This multidisciplinary approach demands the judicious use, not only of HRCT information and histopathology when needed, but also of all non-standardized data available in individual patients, such as BAL, serology tests and biomarkers.
Unfortunately specific biomarkers are yet to be found and less invasive methods to reach lung tissue are far from being established.
Nevertheless recent advances in clinical research allowed the release of new and promising molecules, nintedanib and pirfenidone, two antifibrotic drugs with diverse mechanisms of action that presented efficacy in slowing functional decline and disease progression in IPF patients.
Finally it must be stressed that in IPF the therapeutic strategy should be individualized to each patient, considering, not only the evaluation of potential benefits and risks, but also and most importantly, taking into account the clinical behaviour of the disease.
Corresponding author. carlos.crobalo@gmail.com


