


Non-cystic fibrosis bronchiectasis remains a common and important respiratory disease to date. It is a chronic pathology and consequently the patients usually require continuous treatment.
In recent decades therapies that do not have scientific evidence of their benefits have been commonly used in non-cystic fibrosis bronchiectasis. Cystic fibrosis has provided the experience to extrapolate therapeutic approaches to other bronchiectasis patients. Finally, in the last few years some trials have been carried out specifically in non-cystic fibrosis bronchiectasis which aim to assess the efficacy of some of the treatments which are commonly used but sometimes without clear indication.
This review will discuss the recent results from these trials, namely mucoactive, anti-inflammatory and antibiotic therapy. Several trials are ongoing and we hope they will be able to add clarification to the management of these patients.
As bronquiectasias não-fibrose quística continuam a ser uma doença respiratória comum e importante. Trata-se de uma patologia crónica e, consequentemente, os doentes geralmente precisam de um tratamento contínuo.
Nas últimas décadas, tratamentos sem evidência científica dos seus benefícios foram comumente usadas nas bronquiectasias não-fibrose quística. A fibrose quística serviu de experiência para extrapolar a abordagem terapêutica para outros doentes com bronquiectasias. Finalmente, nos últimos anos, foram realizados alguns ensaios bronquiectasias não-fibrose quística que visam avaliar a eficácia de alguns dos tratamentos que são comummente usados mas por vezes sem uma clara indicação.
Nesta revisão serão apresentados os resultados recentes destes ensaios, nomeadamente sobre o tratamento mucoactivo, anti-inflamatório e antibiótico. Diversos estudos estão a decorrer e esperamos que estes venham a esclarecer a abordagem mais adequada destes doentes.
Bronchiectasis (BE) is an abnormal and irreversible dilation of the bronchi, which has numerous causes. Its frequency depends on the patient's age and sex, social and economic conditions and the degree of applied investigation.
There was little interest in the investigation of non-cystic fibrosis BE, this includes therapeutic approaches, in the last few decades, probably due to supposedly low prevalence and the assumption that treatment is the same for all patients and that little can be done to change the symptoms and evolution.
The publication of diagnosis and treatment reviews in the last few years1,2 demonstrates a growing interest in this pathology. The level of evidence for most recommendations however is low, because of the absence of large double-blind, placebo-controlled trials.3,4
The existence of strong evidence supporting the use of some drugs in patients with cystic fibrosis (CF) does not mean that they will be good for patients with BE of another etiology. So, it is not correct to extrapolate the CF trial results to the non-CF patients.
In CF, the forced expiratory volume in 1s (FEV1) is one of the most important trial end-points. In non-CF BE it has been difficult to establish appropriate end-points to evaluate the effect of new therapeutic interventions. To date it seems that improvement in quality of life is one of the most important outcome measures.5
BE is characterized by a vicious cycle of infection, inflammation and further sputum production. In this review we decided to focus on three important pharmacological groups that aim to interfere with each part of the cycle and in which there have been relevant advances. Nevertheless, BE treatment should be embraced and specific therapies for the underlying cause as well as interventions like physiotherapy, pulmonary rehabilitation, nutritional support and, in selected patients, surgical intervention need to be kept in mind.
Mucoactive therapyRegardless of the cause, BE is mainly characterized by bronchial infection and persistent inflammation which could be the cause and consequence of impaired airway mucous clearance. The mucus progressively becomes viscous due to the presence of inflammatory cells, microorganisms and large polymers and turns into sputum, overwhelming the ciliary clearance capacity.6
The mucus clearance requires a balance between periciliary liquid volume, mucus composition and volume and normal ciliary beat frequency.6 One of these steps could be more particularly affected depending on the cause of BE; the therapeutic intervention should ideally be focused on the main mechanism.7 Unfortunately in many cases this is not clear and there is probably a mix of mechanisms involved so the development of combined therapies would be more appropriate.
The pathogenesis process mostly accepted in CF indicates a relative dehydration and a reduction in airway surface liquid volume.8 Therefore airway hydration is an important goal in the overall therapeutic management of this disease. However, it is generally accepted that even in the absence of dehydration the increase in water improves mucus clearance by decreasing surface interactions.9 In this context the most recent advances in the treatment of mucociliary dysfunction are targeted at increasing hydration on airway surface by inhaled hyperosmolar agents, like mannitol and hypertonic saline (Table 1). Both the agents increase the osmolarity of the airway surface fluid causing influx of water into the airway and reducing the viscoelastic properties of the mucus by breaking some of the mucin bonds.10
As inhalation of hyperosmolar agents may induce airway narrowing and a reduction in FEV1 of about 15% in sensitive subjects, an assessment of bronchial hyperresponsiveness is recommended before starting treatment.11,12
MannitolMannitol is a nonionic sugar alcohol, commonly used as an osmotic agent, which increases mucus clearance. The precise mechanism of this action is unknown.9 Studies have demonstrated that the effect of mannitol is acute rather than cumulative but it has a sustained effect for up to 24h. The mucociliary clearance effect was also found in all regions of the lung, including the peripheral region.13
Mannitol capsules (40mg) which contain dry powder for inhalation using an inhaler device are commercially available.12 They have been approved for the treatment of CF in adults aged ≥18 years. The recommended dose is 400mg (which requires the inhalation of the content of 10 capsules loaded individually into the inhaler) twice a day, once in the morning and once in the evening.12 Mannitol has the advantage of being a powder formulation and therefore has a shorter delivery time which avoids the usual maintenance and cleaning issues related with nebulizer devices.
An international phase-III randomized double-blind placebo-controlled trial of inhaled dry powder mannitol (400mg) was carried out on 324 CF patients, for a 26-week period, twice daily. This showed a significant improvement in FEV1 (change from baseline 118mL (6.5%) versus 26mL (2.4%); p<0.001). Improvements in FEV1 were seen fairly early (at 6 weeks) and were maintained up to 52 weeks. There was also a 35.4% reduction in the incidence of exacerbation on the mannitol group. It is worth noting that the lung function improvement was found irrespective of the concomitant use of recombinant human deoxyribonuclease (rhDNase). Cough and haemoptysis were the most common adverse events. Overall they were mild or moderate and only a small proportion of patients had to discontinue the treatment.14
Given these results, mannitol was also tested on non-CF BE and even though the first studies were carried out on small groups of patients, during short periods of time and mostly relating to non-clinical issues, they showed promising results of its use in this patient population.11
An open-label study of mannitol (400mg), carried out on 9 patients with BE, once daily, during a 12-day period documented a highly significant improvement in quality of life, which is assessed by St George's Respiratory Questionnaire (SGRQ), and maintained for 6–10 days after cessation of treatment. It is important to note that patients reported a reduction in cough during both day and night, a reduction of sputum in the morning and an increase in sleeping time. Some properties of sputum were changed, increasing cough efficacy. No significant changes in lung function were found.9 A phase-3 multicentre randomized controlled trial, available only in abstract, involving 185 BE patients with mild-to-moderate lung function demonstrated an improvement in health-related quality of life with inhaled mannitol (320mg) over 12 weeks. Furthermore a prolonged time free of antibiotics and a lower total use of antibiotics compared with placebo was observed.15
A phase-III clinical trial is in progress and the purpose of this study is to examine the efficacy and safety of a 52-week treatment with 400mg inhaled mannitol, twice daily, against a control group. The primary outcome measure is the effect of mannitol in reducing the pulmonary exacerbations and the secondary outcomes are the difference in quality of life, antibiotic use, number of hospitalizations, sputum volume, daytime sleepiness scores, lung function and health related costs.16
Hypertonic saline solutionHypertonic saline (HS) is an ionic substance quickly transported across the epithelium and delivered using an ultrasonic or jet nebulizer. The highest well tolerated concentration is 7%. Its utility was well documented for CF in a randomized placebo-controlled trial in 164 patients, twice daily, over 48 weeks. The HS group had significantly fewer pulmonary exacerbations, an improvement in quality of life, a reduction in absenteeism from school and work and an improvement in FEV1 of 65ml.17 A Cochrane review in 2009, including 12 trials, concluded that HS is a safe, low-cost and effective therapy in CF.18However, the HS recommendation for non-CF BE is not clear.
In a crossover trial 24 BE patients, who produced less than 10g of sputum per day, were randomly selected to receive four single treatment schedules once a week, for 4 weeks. This included active cycle breathing technique (ACBT) alone; nebulized terbutaline followed by ACBT after 10min; nebulized terbutaline followed after 10min by nebulized isotonic saline (IS) (09%) then ACBT; nebulized terbutaline followed after 10min by nebulized HS (7%) then ACBT. Both nebulized IS and HS were significantly more effective in increasing sputum yield, reducing sputum viscosity and improving ease of expectoration but HS was significantly better than IS.19
In another randomized single blind crossover trial to evaluate IS and HS daily for 3 months in 28 BE patients, a significant improvement in lung function (FEV1 and FVC) and quality of life was documented by changes in global scores and subscales of symptoms and impact of SGRQ in favor of HS.20
A recently published research showed different results. Forty patients were randomized to inhale IS or HS 6% daily for 12 months and no important clinical superiority of HS over IS was identified. Nevertheless, there were significant improvements in quality of life, lung function and sputum colonization in both groups. Noteworthy is that prior to the trial 85% of the patients regularly performed airway clearance techniques, which means that the daily use of a saline solution may offer additional benefits.21
Even though there is some evidence favoring HS, it seems that both hypertonic and isotonic saline are beneficial in non-CF BE.19,21,21 These results agree with another randomized placebo-controlled study that evaluated the clinical utility of long-term humidification therapy in 108 patients with COPD (n=63) or BE (n=45). They used fully humidified high flow air at 37°C through nasal cannulae, daily, over a 12-month period. The patients on long-term humidification therapy showed significantly fewer exacerbation days, increased time to first exacerbation, improvement in quality of life and lung function. There was a non-significant reduction in exacerbation frequency.22
N-acetylcysteineN-acetylcysteine (NAC) is commonly used in the treatment of BE patients. It is a mucolytic agent that disrupts the disulfide bonds in mucus when inhaled. After oral administration there is no NAC in airway secretions but cysteine is detected in the plasma and induces an increase in glutathione levels in the plasma and lung which has antioxidant properties. Therefore the benefits of this agent may come from its antioxidant properties and not its mucolytic characteristics. NAC has also antibacterial properties by reducing the ability of bacteria to adhere to epithelial cells.23 Despite this, in 2001 a Cochrane review concluded that there is no evidence to recommend the use of NAC in non-CF BE.24
Sputum clearance is a complex mechanism, with several targets, which is not completely understood. Trials of mucoactive agents are scarce and there are some discordant results. The latter may be explained by the fact that the analyses are usually independent of the underlying disease. With sub-group analyses, clear benefits would probably be discovered, which underlines the importance of defining the etiology. An example of this is the bad results obtained with rhDNase in non-cystic fibrosis BE,25 as it not only failed to improve FEV1 but also the patients got worse.
Anti-inflammatory therapyAs stated before, persistent inflammation is a cardinal feature of BE26; it is part of a vicious cycle which also involves host susceptibility, sputum hypersecretion, airway obstruction and infection.27 Sometimes, even in the absence of existing infection there is continuous neutrophilic infiltration of the airways which suggests dysregulation of immune responses.28 In this setting, it is reasonable to consider anti-inflammatory and/or immunomodulatory therapies as an option in the management of patients with BE.
Although it is a disease which is increasingly recognized and studied, evidence to support the use of anti-inflammatory therapies is still limited even when taking into account drugs with an established anti-inflammatory role in the treatment of other disorders.
MacrolidesApart from their antimicrobial activity, macrolides have been increasingly used in clinical practice for the treatment of a variety of chronic pulmonary diseases because of their anti-inflammatory and immune-modulatory properties.29–32 Although solid evidence to justify long-term macrolide therapy in many of these disorders is still lacking, beneficial effects have been found in small clinical trials of patients with non-CF BE.33–39 There were considerable variations in the study design of many of these trials, including duration, dose and macrolides tested and outcome measures evaluated but most of them showed consistent evidence of a decrease in sputum volume and some reported a decrease in exacerbation frequency. Several larger studies which have recently been completed corroborate these findings. A significant reduction in exacerbation frequency was found in the preliminary results of a trial with 89 patients which compared the use of azithromycin with a placebo40 as well as in another study yet to be published with 117 patients using erythromycin.41 Accordingly, the largest study to date, which has recently been published,42 demonstrated that a 6 months azithromycin treatment significantly decreased the rate of exacerbations requiring antibiotic therapy, which was sustained for 6 months after completion of treatment, and increased the time to the first exacerbation requiring antibiotics. As well as these beneficial effects, however, it is important to realize that side effects such as gastrointestinal upset, raised transaminase levels, cardiac arrhythmias (particularly prolonged QT interval related), auditory impairment, urogenital candidiasis and, lastly, and possibly most importantly, the risk of microbial macrolide resistance, particularly non-tuberculous mycobacteria (NMT), may occur.30,43
Therefore, macrolides cannot be recommended as routine therapy for non-CF BE before further research is carried out, although their use, particularly azithromycin, can be considered in selected patients.4,44 In patients with frequent respiratory exacerbations or continuous symptoms for more than 6 months, particularly if chronically infected with Pseudomonas aeruginosa, a trial of 3–6 months of 3 times per week 250–500mg azithromycin can be done after ruling out NTM infection.4,44 Transaminase monitoring should be performed in the first few weeks of treatment and then, as well as NTM screening, every 6 months if there is evidence of benefit to the patient in terms of quality of life and frequency of exacerbations and the therapy is not discontinued.4 There are no studies to support either the effectiveness or safety of treatments of more than 12 months duration.
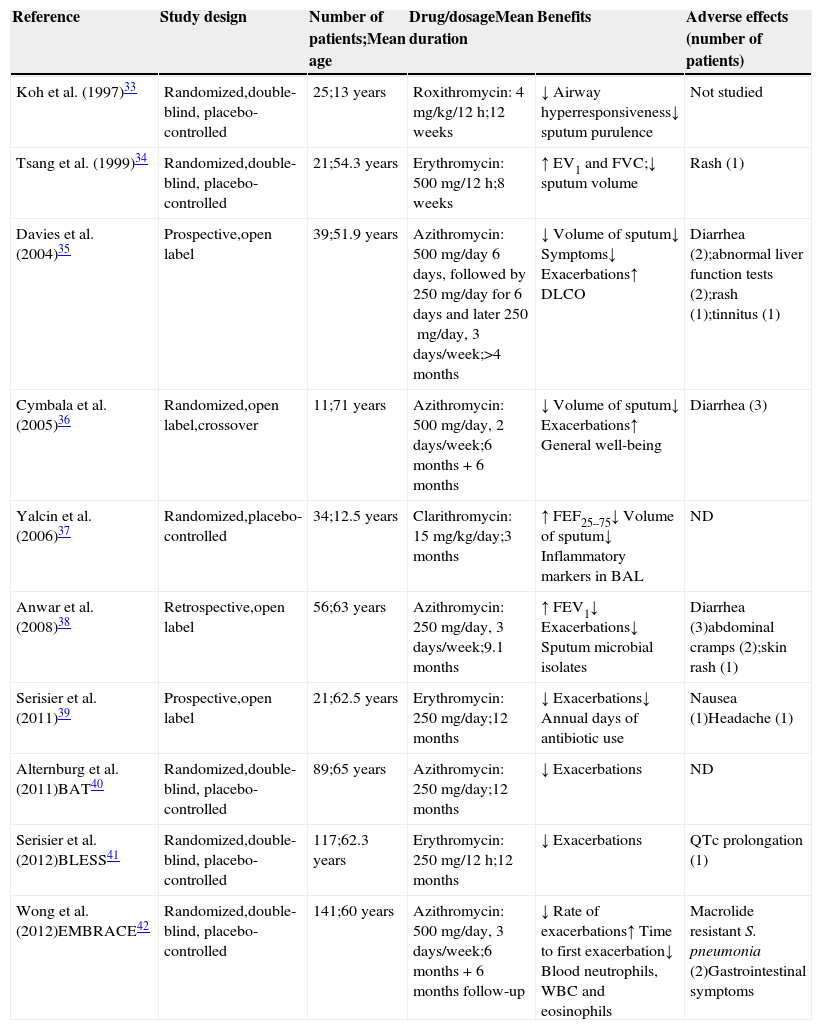
An overview of the available studies showing the effects of macrolides in patients with BE is presented in Table 2.
Clinical trials of macrolide therapy.
| Reference | Study design | Number of patients;Mean age | Drug/dosageMean duration | Benefits | Adverse effects (number of patients) |
|---|---|---|---|---|---|
| Koh et al. (1997)33 | Randomized,double-blind, placebo-controlled | 25;13 years | Roxithromycin: 4mg/kg/12h;12 weeks | ↓ Airway hyperresponsiveness↓ sputum purulence | Not studied |
| Tsang et al. (1999)34 | Randomized,double-blind, placebo-controlled | 21;54.3 years | Erythromycin: 500mg/12h;8 weeks | ↑ EV1 and FVC;↓ sputum volume | Rash (1) |
| Davies et al. (2004)35 | Prospective,open label | 39;51.9 years | Azithromycin: 500mg/day 6 days, followed by 250mg/day for 6 days and later 250mg/day, 3 days/week;>4 months | ↓ Volume of sputum↓ Symptoms↓ Exacerbations↑ DLCO | Diarrhea (2);abnormal liver function tests (2);rash (1);tinnitus (1) |
| Cymbala et al. (2005)36 | Randomized,open label,crossover | 11;71 years | Azithromycin: 500mg/day, 2 days/week;6 months+6 months | ↓ Volume of sputum↓ Exacerbations↑ General well-being | Diarrhea (3) |
| Yalcin et al. (2006)37 | Randomized,placebo-controlled | 34;12.5 years | Clarithromycin: 15mg/kg/day;3 months | ↑ FEF25–75↓ Volume of sputum↓ Inflammatory markers in BAL | ND |
| Anwar et al. (2008)38 | Retrospective,open label | 56;63 years | Azithromycin: 250mg/day, 3 days/week;9.1 months | ↑ FEV1↓ Exacerbations↓ Sputum microbial isolates | Diarrhea (3)abdominal cramps (2);skin rash (1) |
| Serisier et al. (2011)39 | Prospective,open label | 21;62.5 years | Erythromycin: 250mg/day;12 months | ↓ Exacerbations↓ Annual days of antibiotic use | Nausea (1)Headache (1) |
| Alternburg et al. (2011)BAT40 | Randomized,double-blind, placebo-controlled | 89;65 years | Azithromycin: 250mg/day;12 months | ↓ Exacerbations | ND |
| Serisier et al. (2012)BLESS41 | Randomized,double-blind, placebo-controlled | 117;62.3 years | Erythromycin: 250mg/12h;12 months | ↓ Exacerbations | QTc prolongation (1) |
| Wong et al. (2012)EMBRACE42 | Randomized,double-blind, placebo-controlled | 141;60 years | Azithromycin: 500mg/day, 3 days/week;6 months+6 months follow-up | ↓ Rate of exacerbations↑ Time to first exacerbation↓ Blood neutrophils, WBC and eosinophils | Macrolide resistant S. pneumonia (2)Gastrointestinal symptoms |
BAT: Bronchiectasis and long-term Azithromycin Treatment; BLESS: Bronchiectasis and Low-dose Erythromycin Study; EMBRACE: Effectiveness of Macrolides in patients with BRonchiectasis using Azithromycin to Control Exacerbations.
Despite their potent anti-inflammatory action, long-term systemic corticosteroids cannot be used due to potential adverse effects. Inhaled corticosteroids, however, have been studied in patients with BE and a 2009 review by Kapur et al.45 identified six randomized clinical trials which enrolled 278 of 303 randomized adult patients. This meta-analysis concluded that there was insufficient evidence to recommend the routine use of inhaled steroids in adults with stable state BE but considered that a therapeutic trial might be justified in patients with difficult to control symptoms; these would have to be closely monitored for adverse events especially if high doses were used.
A recently published Spanish study46 reported the potentially beneficial effect of the addition of a long acting beta-2 adrenergic to inhaled corticosteroid on clinical parameters and health-related quality of life by allowing the dose of the inhaled steroid to be reduced to half with a reduction of local side effects.
These findings need to be assessed with larger controlled randomized trials.
Other anti-inflammatory agentsNon-steroidal anti-inflammatory drugs (NSAID) may also be a potentially attractive therapy in patients with BE as ibuprofen has demonstrated beneficial effects on people with CF.47,48 However, two Cochrane Database systemic reviews searching for use of oral49 and inhaled NSAID50 in non-CF BE only identified one single trial of inhaled indomethacin versus placebo in 24 adults, 8 of them with BE. This study documented a significant reduction in sputum production and dyspnea in the treatment group over 14 days but further studies on the efficacy of NSAIDs in treating patients with BE are needed.
Like macrolides, another class of drugs that have recognized anti-inflammatory and immunomodulatory properties are hydroxy-methyl-glutaryl-conzymeA reductase inhibitors (i.e. statins) which have demonstrated in vitro inhibition of neutrophil migration and epithelial cell production of chemoattractants and proteases and potentiation of macrophage efferocytosis.51,52 As efferocytosis appears to be involved in the pathogenesis of a variety of chronic lung and systemic inflammatory disease including BE, this would seem an attractive target for statins therapy.53 There are currently ongoing clinical trials to evaluate the effect of atorvastatin on patients with BE, with and without P. aeruginosa infection.54,55
Leukotriene receptor antagonists may theoretically be of benefit as they inhibit neutrophilic mediated inflammation but there are no controlled studies to date to support their use in BE.56
Methylxanthines are also theoretically of use in BE as they are purported to have anti-inflammatory properties but, although there are no published studies to date,57 there are trials currently underway to assess the effect of theophylline in the treatment of bronchiectasis.58,59
Agents specifically targeting a particular mediator of the immune response might be an interesting new class of drugs in the future. Roflumilast and specific monoclonal antibodies, e.g. against IL-8, matrix metalloproteases (MMPs) or neutrophil elastase, are in this group but the safety and tolerability of these drugs still need to be assessed in phase II and III studies.
Antibiotic therapyBE provides the perfect environment for colonization by various microorganisms, as mucociliary clearance is impaired, facilitating rapid bacterial growth on the airways mucosal surface without tissue invasion.
In spite of the fact that these bacteria do not cause invasive disease and are usually less virulent than those that invade nearby tissues, they are able to trigger an inflammatory response that aims to eliminate the microorganism; when this purpose fails, however, the inflammation becomes chronic and leads to progressive severe lung injury.
These pathogens can also develop the means to facilitate their own survival, overcoming host defence mechanisms and antimicrobial actions through biofilm production and other bacterial resistance mechanisms. Thus, the chronic colonization process that occurs in the respiratory tract of patients with BE is called “pathogenic colonization”60 and can be divided into three different phases: initial colonization (the first isolate of a microorganism), intermittent colonization (intermittent isolation of the same microorganism in cultures separated by, at least, a month, representing a chronic colonization process with a low bacterial load) and chronic colonization (three or more successive positive cultures for the same microorganism with, at least, one month apart, within a period of six months).60
Exacerbation is a clinical situation that can happen during any of the three scenarios mentioned above, and it has the potential to worsen lung function deterioration.60 The diagnosis of exacerbation in these patients is particularly difficult and is based on the acute development of manifestations such as changes in sputum characteristics (increased volume, purulence and viscosity), haemoptysis, breathlessness, worsening of cough, chest pain, fever, asthenia, anorexia, weight loss, physical changes in thoracic examination, desaturation, decline in lung function, radiologic signs of lower respiratory tract infection and elevation of systemic biomarkers of inflammation.4
Several microorganisms can be isolated from the respiratory tract of patients with non-CF BE and the acquisition and clearance of a strain is a complex, dynamic process involving host factors and receptor sites on the organism that may help define its ability to persist and damage airways. There is a slight difference in the dynamics of colonization according to age.
The most frequently isolated pathogens are Haemophilus influenzae, P. aeruginosa and Streptococcus pneumonia.61,62 In children the predominant isolated pathogen is H. influenzae,60–65 whereas in adults, although H. influenzae is still the most frequently isolated pathogen,26,28,61 there is a significantly higher isolation rate of P. aeruginosa.62S. pneumoniae and Moraxella catharralis are also found in a significant but variable rate and Aspergillus fumigatus is rarely isolated.62 Isolation of Staphylococcus aureus should lead to consideration of underlying CF.3,26,66
P. aeruginosa has a propensity to persist in bronchiectatic airways due to its capacity to produce virulence factors and modulate immune defences by quorum signaling and biofilm production.67
Since infection (and particularly P. aeruginosa infection) plays a major role in causing and perpetuating BE, reducing the microbial load using antibiotics is a cornerstone of therapy,68 either through treating exacerbations or by using suppressive antibiotic strategies.
However, the isolation of one or more pathogens in the sputum of patients with non-CF BE is not necessarily an indication for antibiotic treatment, particularly in adults62; there are three main criteria for prescribing antibiotics in this setting: exacerbation, eradication of the first isolate of P. aeruginosa and suppressive therapy in steady-state BE chronically colonized with P. aeruginosa.
Treatment of exacerbationsEarly treatment of exacerbations is particularly important as it could probably limit the vicious circle of infection/inflammation that is a determinant factor of lung damage.
The management of these patients includes routine cultures of respiratory secretions to identify infecting organisms and guide antibiotic selection as these patients frequently carry the same bacteria for prolonged periods of time. The current recommendation is to perform cultures every three months on average, so that updated information is available to guide treatment when a pulmonary exacerbation occurs.4 Without this information the choice of antibiotic has to be empirical, based on the most common organisms associated with exacerbations in these patients, in particular P. aeruginosa. Nevertheless, before starting antibiotic therapy, a sputum sample should be sent off for culture, after which antibiotics should be started and then eventually changed in accordance with the microorganism isolated and its antibiogram. However, in vitro antibiotic sensitivity does not always correlate with therapeutic response, which happens with microorganisms able to form a biofilm, where the activity of many antibiotics is diminished.69 So, although an antibiogram is essential for guiding treatment, the value of conventional in vitro susceptibility testing for these patients is being questioned.
It is also common to isolate more than one microrganism or distinct morphotypes of the same pathogen with the same or different patterns of antimicrobial sensitivity. Selecting an antibiotic combination that covers all the isolates can be difficult without resorting to an impractically large number of antibiotics.
The route of administration will depend on the severity of the exacerbation and the evidence of colonization by multidrug resistant microorganisms.
Mild exacerbations can be treated orally with a fluoroquinolone on an outpatient basis.70,71 The addition of inhaled tobramycin solution (TS) to ciprofloxacin was studied because of concerns regarding virulence and potential resistance of Pseudomonas. In a double-blind, multicenter trial, 53 patients with non-CF BE and respiratory exacerbations due to Pseudomonas were randomly assigned to receive ciprofloxacin plus TS or plus placebo for two weeks.72 The addition of TS did not improve clinical outcomes although there was a marked reduction of Pseudomonas density in the sputum. Based on this, TS cannot be recommended alone or in combination with ciprofloxacin for acute exacerbations in non-CF BE.
Intravenous therapy is reserved for oral therapy failure, severe exacerbations or microorganisms resistant to oral antibiotics. The most commonly selected intravenous regimen combines two antibiotics (usually a β-lactam anti-Pseudomonas: piperacillin-tazobactam, ceftazidime or meropenem) and an aminoglycoside.60,70 The rationale for choosing two rather than one is based on the possibility of obtaining synergic effects and decreasing the risk of antibiotic resistances. Regarding the aminoglycoside, tobramycin is recommended due to its strong activity against P. aeruginosa compared to other aminoglycosides. In cases of tobramycin resistance, amikacin can be used.
Adding an inhaled antibiotic to an intravenous one has not been shown to provide clinical benefits.73
There is no evidence to recommend combination antibiotics in patients colonized with microorganisms other than P. aeruginosa, unless there is more than one pathogen.
Exacerbations should be treated using a high dosage targeted antibiotic therapy,3,4 due to the fact that the volume of distribution and total body clearance for hydrophilic drugs (such as aminoglycosides, penicillins, and cephalosporins) are increased because these patients are generally undernourished and have decreased adipose tissue.
The duration of antibiotic therapy is also a matter of debate requiring further investigation but the expert consensus is that 14 days should be recommended for all exacerbations.3,4,60,69
Treatment of Pseudomonas aeruginosa infection/colonizationAs the disease progresses chronic infection by P. aeruginosa becomes common and it seems to be an independent risk factor for accelerated loss of pulmonary function and decreased survival. Conversion of P. aeruginosa to the mucoid phenotype worsens prognosis although it is more common in patients with CF.74,75
Although there are no studies to guide practice following the first isolation, an attempt to eradicate seems pragmatic for what it should be treated aggressively regardless of the patient's clinical signs or symptoms in order to eliminate the microorganism from the sputum as it is very difficult to do so once chronic colonization is established.3,4
In non-CF BE the strategy proposed for eradication is oral ciprofloxacin for 3 weeks and if it fails, the same protocol as for CF is recommended (nebulized antibiotic: tobramycin, sodium colistimethate or aztreonam lysine) plus oral ciprofloxacin for 3 weeks, followed by inhaled antibiotics for 3–12 months.4,60,76 An alternative would be an association of 2 anti-Pseudomonas intravenous antibiotics for 14–21 days, followed by an inhaled antibiotic for 3–12 months.4,60,76 As was stated above, an antibiogram is essential for guiding treatment although it is recognized that in vitro sensitivity does not always correlate with therapeutic response. However, for nebulized antibiotics the interpretation of the antibiogram should take into consideration that, through this form of delivery, antibiotic achieves much higher concentrations in the bronchial mucosa overcoming the mechanisms of resistance.77
The persistence of bacteria despite aggressive treatment is thought to be due to several factors such as poor penetration of antibiotics into purulent airway secretions, native or acquired antibiotic resistance and production of biofilms by the bacteria that may render antibiotics ineffective or interfere with host defences.
To prevent the decline in lung function associated to chronic bronchial colonization with this pathogen, nebulized antibiotics that show in vitro activity against P. aeruginosa are frequently used as chronic suppressive therapy. The aim of this strategy is to reduce the bacterial burden (as in this phase it is virtually impossible to eradicate the pathogen) and, thus, reduce the inflammatory response that leads to structural lung damage and loss of function,4,78 avoiding the cumbersome and toxic iatrogenic effects associated to oral or intravenous route.79 It seems that this strategy may reduce the frequency and severity of respiratory exacerbations and the decline in lung function.80,81
One trial conducted in 2000 randomly assigned 74 patients with non-CF BE and P. aeruginosa infection to receive aerosolized tobramycin (300mg, twice daily) or aerosolized placebo for 28 days.80 Patients in the treatment group demonstrated a 10,000-fold reduction in Pseudomonas density, but no change in FEV1 as compared to controls. The same results were attained in a similar trial conducted in 2001.82
In 2005, a 6 month double blind crossover study of aerosol tobramycin was carried out on 30 patients and revealed no change in exacerbation rate, but the number of hospitalizations and duration of hospital stay were reduced during the tobramycin phase.83
In 2005 a smaller uncontrolled study was carried out in which aerosolized tobramycin (300mg, twice daily) was administered to 41 patients with non-CF BE and Pseudomonas infection; the protocol alternated two weeks with therapy and two weeks without, for a total of 12 weeks.84 Treatment was associated with a decrease in symptoms and improvements in health-related quality of life (QOL). However, 10 of 41 patients were unable to complete the protocol because of side effects (cough, wheezing or worsened dyspnea), and two of the patients who completed the trial acquired tobramycin-resistant Pseudomonas species.
In 2007 an uncontrolled study examined the efficacy of nebulized colistin in a mixed population of patients with COPD and bronchiectasis colonized with PSAE and showed an improvement in quality of life and slower decline with FEV1 with treatment.85
Currently, many other clinical trials are being carried out with the purpose of establishing the indications for inhaled antibiotic therapy in these patients and define which antibiotics to use and the most appropriate devices to deliver them.86
In fact, at the moment, the clinical evidence that supports the use of chronic inhaled antibiotherapy in non-CF BE, chronically colonized with Pseudomonas, is scarce but all the data suggest that this therapeutic strategy seems to control the symptoms, prevent the progression of the disease and reduce the morbidity with no relevant adverse events.87,88
The chronic antibiotherapy may be intermittent (28 days on, 28 days off) when using tobramycin or aztreonam lysine or continuous when using sodium colistimethate. However, continuous inhaled antibiotherapy should be prescribed using 2 different antibiotics alternately in patients that are very symptomatic in off-periods, or with severe lung function impairment or recurrent pulmonary exacerbations despite taking one antibiotic every other month.4
Nevertheless, there is still a degree of uncertainty about when to prescribe the various inhaled antibiotics and what to choose.
At the moment the recommendations3 are to treat patients with non-CF BE chronically colonized with P. aeruginosa and with frequent acute exacerbations (three or more per year) or progressive deterioration of lung function, with inhaled antibiotics; it seems reasonable to use tobramycin as the first choice because of the extensive information supporting its efficacy and good safety record after many years of use. The level of evidence and clinical benefit is greatest for patients with moderate or severe lung disease.
The option for inhaled aztreonam instead of tobramycin may be considered if there is evidence of inhaled tobramycin intolerance, clinical deterioration despite inhaled tobramycin, a strong possibility of increasing adherence due to the fact that inhaled aztreonam can be delivered more rapidly than tobramycin and evidence or possibility of pregnancy when aminoglycosides are relatively contraindicated. Data are needed from well-designed comparative effectiveness trials for tobramycin and aztreonam to provide for a more informed decision as to which to use.
Given the less than robust data supporting its use and the suggestion that it may be inferior, it seems reasonable to prescribe colistin only in cases of tobramycin therapeutic failure or intolerance.
In conclusion, controlling respiratory infection/colonization is the cornerstone of therapy in non-CF BE with a significant impact on survival for which more robust data are needed to support decisions in this area.
ConclusionBE has recently became a hot topic. First results of well designed trials have been published and several studies are in progress. However, there are many questions regarding treatment which are still waiting for scientific answers.
It will be important to design studies involving a greater number of patients, with different levels of severity, more specific aetiological information and carried out over longer time periods to clarify the real benefits of these promising therapies and bring new insights into this interesting topic.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.


