




Five years survival of lung cancer is 16%, significantly lower than in prostate (99.9%), breast (88.5%) and colon (64.1%) carcinomas. When diagnosed in the surgical stage it increases to 50% but this group only comprises 14–16% of the cases. DNA methylation has emerged as a potential cancer-specific biomarker. Hypermethylation of CpG islands located in the promoter regions of tumour suppressor genes is now firmly established as an important mechanism for gene inactivation.
This retrospective study included 40 squamous cell carcinomas and 40 adenocarcinomas in various surgical TNM stages to define methylation profile and possible silencing of DNA repair genes – MLH1 and MSH2 – using Methylation-Specific PCR and protein expression by immunohistochemistry in tumoural tissue, preneoplastic lesions and respiratory epithelium with normal histological features.
The protein expression of MLH1 and MSH2 genes, in the available preneoplastic lesions and in normal cylindrical respiratory epithelium appeared reduced. The frequency of promoter hypermethylation found on these DNA repair genes was elevated, with a higher prevalence of methylation of MLH1 gene in 72% of squamous cell carcinoma. The differences are not so obvious for MSH2 promoter hypermethylation. No correlation was found among the status of methylation, the protein expression and the clinicopathological characteristics.
With a larger study, a better characterization of the hypermethylation status of neoplastic and preneoplastic lesions in small biopsies would be achieved, inherent to tumour histology, heterogeneity and preservation, and finally differences in the study population to elucidate other possible mechanisms of altered expression of the hMLH1 and hMSH.
A sobrevivência aos cinco anos no cancro do pulmão é de 16%, significativamente inferior que nos carcinomas na próstata (99,9%), mama (88,5%) e cólon (64,1%). Quando diagnosticado na fase cirúrgica aumenta até 50%, mas este grupo é apenas constituído por 14-16% dos casos. A metilação do ADN surgiu como um potencial marcador biológico específico do cancro. A hipermetilação das ilhas CpG localizadas nas regiões promotoras de genes supressores do tumor está agora firmemente estabelecida como um mecanismo importante para a inativação do gene.
Este estudo retrospetivo incluiu 40 carcinomas das células escamosas e 40 adenocarcinomas em vários estádios cirúrgicos TNM para definir o perfil da metilação e o possível silenciamento de genes de reparação do ADN - MLH1 e MSH2 - usando metilação PCR específica e expressão da proteína por imuno-histoquímica no tecido tumoral, lesões pré-neoplásicas e epitélio respiratório com características histológicas normais.
A expressão da proteína dos genes MLH1 e MSH2, nas lesões pré-neoplásticas disponíveis e no epitélio respiratório cilíndrico normal, pareceu reduzida. A frequência da hipermetilação promotora encontrada nestes genes reparadores de ADN foi elevada, com uma maior prevalência da metilação do gene MLH1 em 72% de carcinoma de células escamosas. As diferenças não são tão óbvias para a hipermetilação do promotor MSH2. Não foi encontrada correlação entre o estado de metilação, a expressão da proteína e as características clínico-patológicas.
Com um estudo mais amplo, seria alcançada uma melhor caracterização do estado da hipermetilação das lesões neoplásicas e pré-neoplásicas em pequenas biopsias, inerente à histologia, heterogeneidade e preservação do tumor, e, finalmente, às diferenças na população estudada para elucidar outros mecanismos possíveis da expressão alterada do hMLH1 e hMSH.
Epigenetics of human cancer has been overshadowed by human cancer genetics since 1983. Increasingly visible with a growing understanding of specific epigenetic mechanisms and their role in cancer,1 the modifications refer to a number of molecular mechanisms that regulate gene expression without changing DNA sequence2: (1) alteration of methylation status of DNA within CpG islands (the main human epigenetic modification)3; (2) covalent modification of histone tails; (3) gene regulation by micro-RNA (miRNA).
While early embryonic cells lack methylation (as it is not transmitted via the germline), methylation is essential for the development and regulation of gene expression and controls expression of oncofetal genes in postnatal life by imprinted genes and tissue-specific gene expression.2 This perfect equilibrium in normal cells is transformed in cancer cells. DNA methylation is believed to contribute to cancer initiation and progression by gene inactivation. This can have important consequences if the inactivated genes are essential for the control of normal cell growth, differentiation, or apoptosis.4 The mechanisms that regulate normal and aberrant methylation are neither fully understood nor are the mechanisms of methylation that interfere with transcription.4
The mismatch DNA repair (MMR) system is composed of a few well-conserved proteins. The essential components of MMR system, MutS, MutL, MutH and Uvr, were identified in Escherichia coli. In addition, all eukaryotic organisms, including humans, have MutS homologs and MutL homologs. The MLH1 and MSH2 genes provide instructions for making a protein that plays an essential role in DNA repair. These proteins fix mistakes that are made when DNA is copied (DNA replication) in preparation for cell division. The MLH1 protein joins with another protein, the PMS2, to form an active protein complex. This protein complex coordinates the activities of other proteins that repair errors during DNA replication. The repairs occur by removing the section of DNA and replacing it by a correct DNA sequence. The MSH2 protein joins with one of the two other proteins, the MSH6 protein or the MSH3 protein, to form an active protein complex. This active protein complex identifies places on the DNA where mistakes have been made during DNA replication.
The prognosis of lung cancer is very limited by the difficulties of diagnosing early stage disease amenable to surgery. Only 10% of cases can benefit from local treatment with long-term survival. Despite much progress in the treatment and detection methods of lung carcinoma, the prognosis remains poor. This situation is mainly the result of metastases which are present in more than two-thirds of patients at the time of diagnosis.5,6
The objectives of our study were to characterize the expression of DNA repair proteins MLH1 and MSH2 in tumour tissue, precursor lesions, respiratory epithelia and parenchyma of 80 clinically well-characterized NSCLC patients and also to study the methylation status of two DNA repair genes – MLH1 and MSH2, to correlate between the methylation status of the promoters of MLH1 and MSH2, and their respective protein expression and clinicopathological parameters.
Materials and methodsPatients and tissue samplesAll biopsy and resection specimens were reviewed by the Pathology Department of the University Hospital of Coimbra, to verify non-small cell histology of the lung cancer samples and to determine the histologic subtype. Histological subtypes consisted of 80 cases of surgically staged pulmonary carcinomas as well as corresponding non-neoplastic and pre-neoplastic tissue of adenocarcinomas and squamous cell carcinomas of the lung.
Twenty-four females and 16 males with adenocarcinoma (ADC), and 36 males and 4 females with squamous cell carcinoma (SCC) were confirmed according to the latest World Health Organization Classification of Lung Cancer (2004).7 The average age was 64.9±9.88 years at the time of diagnosis.
The TNM staging was applied according to the 2010 TNM Classification of Malignant Tumours, 7th Edition8: thirty-nine patients had Stage I disease, 19 had Stage II and 9 had stage III.
Tissue microarrays buildingRepresentative areas of carcinoma, preneoplastic lesion and normal bronchial epithelium and parenchyma, were carefully selected from the haematoxylin-eosin slide, and marked respectively in each slide and the each formalin-fixed, paraffin-embedded block of tissue. Tissue microarrays of 3mm were performed in triplicate in tumour and normal respiratory epithelium and in a single core for preneoplastic lesion, included on the same slide.
Analysis of protein expression: immunohistochemistryIHC satins for hMLH1 and hMLH2 were performed on 3μm thick sections cut from paraffin-embedded TMAs blocks. Commercially available antibodies against these markers were used as per manufacturer's protocols. Monoclonal antibodies used were (clone ZM001, 1:200, Zymed) for MLH1and (clone FE11, 1:40, Zymed) for MSH2, using the Streptavidin–Biotin Horseradish Peroxidase method. Epitope retrieval using microwave heat or steam pre-treatment was performed as required. Normal endometrium was used as positive control. The normal staining pattern for both MLH1 and MSH2 is nuclear. Lymphocytes and normal bronchial epithelium were used as internal positive control as follow: Normal expression (>75%/+++); Reduced expression: Negative (≤10%/−) and Positive (>10–50%/+ or 50–75%/++).
Following the explained score it was possible to define two definite groups, the positive when present +++/75% nuclear expression and the group with reduced expression. Staining results were examined without knowledge of the status of the molecular analyses.
Methylation-specific polymerase chain reaction assay for the hMLH1 and hMSH2 genesFor DNA extraction, areas from all the slides corresponding to the principal histological patterns were selected. The tumour-rich areas were manually micro-dissected. Five 10μM sections were cut from the paraffin blocks corresponding to the already selected areas and deparaffined by xylene extraction. The resulting tissue pellets were digested with Proteinase K at 56°C overnight. The genomic DNA was isolated using QIAamp DNA MiniKit according to the manufacturer’ protocol (Qiagen, Valencia, CA). By spectrophotometry (Nanodrop ND1000, Thermo Scientific, USA) the purity and concentration of the samples were verified.
The promoter methylation status of the MLH1 gene of all tumour samples and of the MSH2 gene of tumour samples was determined by chemical treatment with sodium bisulfite and subsequent methylation-specific PCR (MSP) analysis as described.9
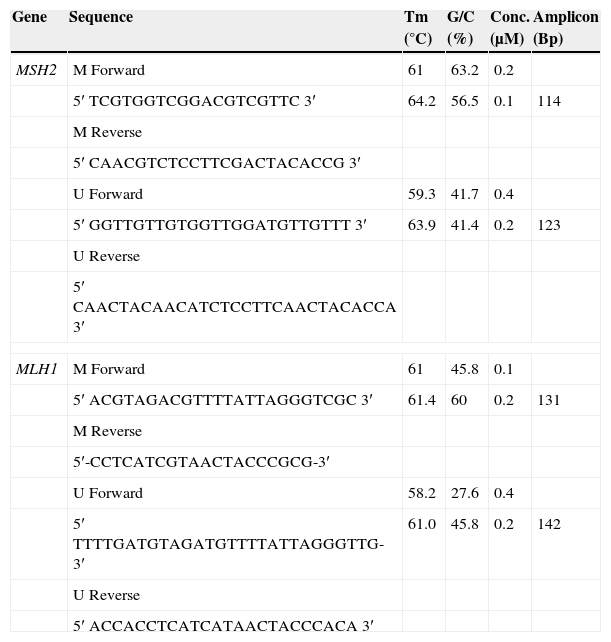
Two sets of primers were specifically designed upstream of the promoter region to discriminate methylated from unmethylated alleles, using the Methprimer Software10 (Table 1).
MLH1 and MSH2 primer concentrations after optimization.
| Gene | Sequence | Tm (°C) | G/C (%) | Conc. (μM) | Amplicon (Bp) |
|---|---|---|---|---|---|
| MSH2 | M Forward | 61 | 63.2 | 0.2 | |
| 5′ TCGTGGTCGGACGTCGTTC 3′ | 64.2 | 56.5 | 0.1 | 114 | |
| M Reverse | |||||
| 5′ CAACGTCTCCTTCGACTACACCG 3′ | |||||
| U Forward | 59.3 | 41.7 | 0.4 | ||
| 5′ GGTTGTTGTGGTTGGATGTTGTTT 3′ | 63.9 | 41.4 | 0.2 | 123 | |
| U Reverse | |||||
| 5′ CAACTACAACATCTCCTTCAACTACACCA 3′ | |||||
| MLH1 | M Forward | 61 | 45.8 | 0.1 | |
| 5′ ACGTAGACGTTTTATTAGGGTCGC 3′ | 61.4 | 60 | 0.2 | 131 | |
| M Reverse | |||||
| 5′-CCTCATCGTAACTACCCGCG-3′ | |||||
| U Forward | 58.2 | 27.6 | 0.4 | ||
| 5′ TTTTGATGTAGATGTTTTATTAGGGTTG-3′ | 61.0 | 45.8 | 0.2 | 142 | |
| U Reverse | |||||
| 5′ ACCACCTCATCATAACTACCCACA 3′ | |||||
The bisulfite modification was performed using the Epitect Bisulfite Kit (Qiagen Valencia, CA) to convert all unmethylated cytosines to uracils, while leaving methylated cytosines unaffected (Table 2).
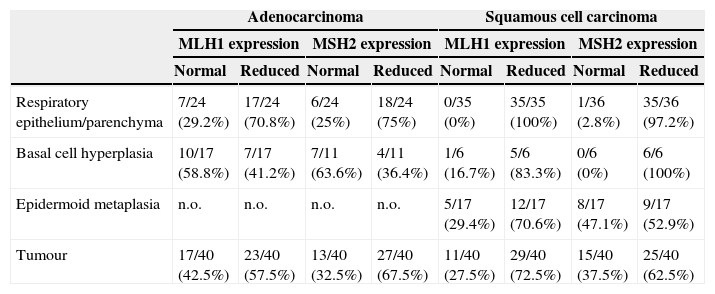
MLH1 and MSH2 positive immunohistochemistry expression in carcinogenesis progression achieved in tissue microarrays.
| Adenocarcinoma | Squamous cell carcinoma | |||||||
|---|---|---|---|---|---|---|---|---|
| MLH1 expression | MSH2 expression | MLH1 expression | MSH2 expression | |||||
| Normal | Reduced | Normal | Reduced | Normal | Reduced | Normal | Reduced | |
| Respiratory epithelium/parenchyma | 7/24 (29.2%) | 17/24 (70.8%) | 6/24 (25%) | 18/24 (75%) | 0/35 (0%) | 35/35 (100%) | 1/36 (2.8%) | 35/36 (97.2%) |
| Basal cell hyperplasia | 10/17 (58.8%) | 7/17 (41.2%) | 7/11 (63.6%) | 4/11 (36.4%) | 1/6 (16.7%) | 5/6 (83.3%) | 0/6 (0%) | 6/6 (100%) |
| Epidermoid metaplasia | n.o. | n.o. | n.o. | n.o. | 5/17 (29.4%) | 12/17 (70.6%) | 8/17 (47.1%) | 9/17 (52.9%) |
| Tumour | 17/40 (42.5%) | 23/40 (57.5%) | 13/40 (32.5%) | 27/40 (67.5%) | 11/40 (27.5%) | 29/40 (72.5%) | 15/40 (37.5%) | 25/40 (62.5%) |
n.o., not observed; normal expression 75%/+++; reduced expression positivity below 75%/−.
Modified DNA was amplified in a total volume of 110μL containing 1× NH4 buffer, 2mM of MgCl2, 200μM of dNTPs, 1U Taq DNA Polymerase (Bioline), different primer pair concentrations were used after optimization (Table 1), considering that we are working with FFPE tissue, an optimization procedure confers better results. PCR was performed for 40 cycles with annealing temperatures of 61°C for 30s and primer extension at 72°C for 60s using 50ng bisulfite-modified DNA. All PCRs were performed with positive controls for both unmethylated and methylated alleles and no DNA control. DNA of healthy human lymphocytes served as positive control for methylated and unmethylated MLH1 and MSH2 state. The first was treated with Methyltransferase (CpG Methyltransferase M.SssI) (New England Biolabs), and subsequently submitted to sodium bisulfite transformation (Epitect Bisulfite, Qiagen), the second was only modified with sodium bisulphite. The CpG Methyltransferase, M.SssI, methylates all cytosine residues (C5) within the double-stranded dinucleotide recognition sequence 5′…CG…3′.
The PCR products were separated on a 4% agarose gel containing ethidium bromide and visualized under UV light, and its size estimated by comparison with a DNA Molecular Weight Marker XIII (50bp ladder) (Roche).
Statistical analysisThe association between clinicopathological variables (pTNM, gender, age) and frequency of methylation and protein expression silencing among the tumour subtypes was assessed using the Pearson χ2 and Fischer's exact test. All tests were two-sided and the level of significance was set at p<0.05.
ResultsProtein expression of MLH1 and MSH2 in tissue microarraysWe investigated MLH1 and MSH2 expression using immunohistochemistry analysis in tissue microarrays comprising 80 primary NSCLC patients, that were respiratory epithelium, preneoplastic lesion (basal cell hyperplasia, metaplasia, dysplasia) and tumoural patterns. Following the explained score it was possible to define two definite groups, normal expression with >75%/+++ nuclear expression and the group with reduced expression <75%/++. After applying these scores, the internal control of lymphocytes had normal expression (data not shown) but tumour adjacent respiratory epithelium and parenchyma were considered reduced for both MLH1 and MSH2. Twenty-three cases (57%) of ADC for MLH1 expression and 27 cases (67%) for MSH2, had reduced expression; in SCC 29 cases (72%) for MLH1 expression and 25 cases (62%) for MSH2 expression had reduced expression.
Co-reduction of both MLH1 and MSH2 was found in 38 cases (19 ADC and 19 SCCs), according with Table 5, probably due to different altered epigenetic or/and genetic mechanisms involved in down-regulation of gene expression. Tumour cells that exhibited an absence of nuclear staining in the presence of non-neoplastic cells and infiltrating lymphocytes with nuclear staining were considered to have an abnormal pattern.
In normal respiratory epithelium reduced expression was found in 70% MLH1 and 75% MSH2 in ADC, and 100% MLH1 and 97% in MSH2 in SCC, to study this we would need cancer-free controls to explain the possible time required to acquire the additional genetic and epigenetic changes that promote tumour progression.
Basal cell hyperplasia in cases of adenocarcinoma presented normal expression of MLH1 and MSH2. Squamous cell carcinomas revealed reduced expression of MLH1 and MSH2 in 72% and 62.5% respectively. Fig. 1 represents the IHC expression heterogeneity.
 basel cell hyperplasia, (B) acinar pattern with reduced expression (−), (C) bronchioloalveolar pattern with normal expression (+++) and (D) solid pattern with reduced expression (++); MSH2 IHC expression in epidermoid carcinoma in situ and (E) corresponding epidermoid carcinoma (F) 200×.")
MLH1 IHC expression in one mixed type adenocarcinoma, (A) basel cell hyperplasia, (B) acinar pattern with reduced expression (−), (C) bronchioloalveolar pattern with normal expression (+++) and (D) solid pattern with reduced expression (++); MSH2 IHC expression in epidermoid carcinoma in situ and (E) corresponding epidermoid carcinoma (F) 200×.
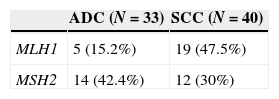
We examined the promoter hypermethylation status of MLH1 and MSH2 genes in 40 squaumous cell carcinoma and 33 adenocarcinoma using MSP assay in manually microdissected tissue. 40 DNA samples of squamous cell carcinoma and 33 samples of adenocarcinoma were analyzed (Table 3).
Clinicopathological distribution of gene promoter methylation and loss of expression at tumour type in 80 patients. Patient's age ranging 64.9±9.88 years (63.4 years – adenocarcinoma 66.55 years – squamous cell carcinoma), 28 females and 52 males.
White grey denotes unmethylated and normal protein expression respectively, dark grey methylated promoter genes. M, male; F, female; ADC, adenocarcinoma; SCC, squamous cell carcinoma; T, tumour size; N, local metastasis; M, distant metastasis; n.a., not available; n.d., not determined.
We observed two patterns of hypermethylation; the partial pattern was the most prevalent (58.33%) for the MLH1 gene (M-U) while for the MSH2 gene the hypermethylation fully pattern (M-M) was the one most verified (76.9%).
MLH1 methylation pattern seems to vary substantially by histological type (Table 3). The prevalence of MLH1 promoter hypermethylation was higher in squamous cell carcinoma (47.5%), with a correlation (p=0.003) between the histological subtype and the methylation status of the promoter of MLH1 gene. The difference is not so obvious in MSH2 promoter hypermethylation; however adenocarcinoma has the highest number of cases (42.4%). No correlation was found between histological subtype and methylation status of MSH2 (p=0.2699).
According to the relationship between the frequency of gender and histological subtype, women with adenocarcinoma as well as men with squamous cell carcinoma have a higher percentage of hypermethylation of MLH1 and MSH2 promoters (Table 4).
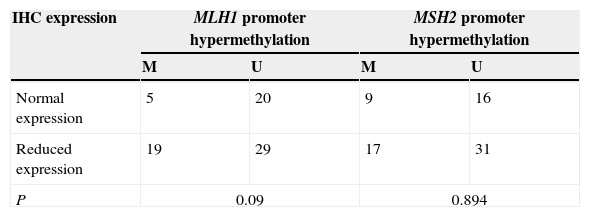
Relationship between the methylation status and IHC protein expressionAccording to the distribution of gene promoter methylation and loss of expression at tumour type in 80 patients, no statistical correlation was observed between the promoter methylation and expression levels in tumour samples, p=0.09 for MLH1 and p=0.894 for MSH2 (Tables 5–7).
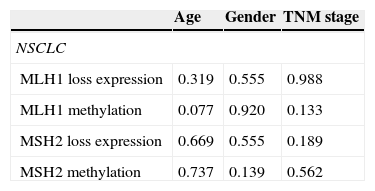
Correlation of MLH1 and MSH2 methylation levels and respective protein expression in relation to the clinicopathological variables of NSCLC tumours (age, gender and TNM stage).
| Age | Gender | TNM stage | |
|---|---|---|---|
| NSCLC | |||
| MLH1 loss expression | 0.319 | 0.555 | 0.988 |
| MLH1 methylation | 0.077 | 0.920 | 0.133 |
| MSH2 loss expression | 0.669 | 0.555 | 0.189 |
| MSH2 methylation | 0.737 | 0.139 | 0.562 |
Moreover, no correlation was found between MLH1 and MSH2 methylation levels and any of the clinicopathological characteristics considered (age, gender, TNM stage and tumour histological type) (Table 7).
We observed two patterns of hypermethylation; the partial pattern was the most prevalent (58.33%) for the MLH1 gene (M-U) while for the MSH2 gene the hypermethylation fully pattern (M-M) was the one most verified (76.9%).
DiscussionMost clinical studies, have recorded moderate to high elevation of MSH2 expression in sporadic tumours like colon, prostate or bladder while there is substantial reduction to near loss of functional MLH1 or MSH2 protein expression in ovarian, lung and stomach tumours relative to their matched normal tissues.11–13 Reduced expression of MLH1 and MSH2 was observed in respiratory epithelia, metaplasia and dysplasia. We know that cancer is a disease involving multiple pathways and genetic lesions, which are necessary for a tumour to become fully established. The story is the same for epigenetic lesions. It is difficult to explain the differential rates of progression of premalignant lesions and differences in behaviour of morphologically similar lesions. Heterogeneity for microsatellite instability (MSI) and promoter methylation in driving these phenomena forward may explain this; however, no previous analysis has examined this in detail.
Alterations in expression of MMR proteins MLH1 and MSH2 have been reported in a variable proportion of NSCLC but very few studies have investigated the role of reduced protein expression in precursor lesions of NSCLC or have investigated their potential prognostic significance in invasive carcinomas. More recent studies have identified changes in methylation in the oral epithelium of smokers and linked these changes to bronchial epithelium.14,15
We found differences concerning the pattern of methylation of MLH1 gene depending on the histological typing of NSCLC (p=0.003) and within the subtype. Fifteen percent of the ADC cases showed hypermethylation of MLH1 compared with 47.5% cases of methylated SCC. Although we found no significant differences in the methylation pattern of MLH1 and MSH2 genes in SCC, ADC has a higher frequency of MLH1 methylated rather than MSH2.
In patients with ADC we also found basal cell hyperplasia that presents MLH1 and MSH2. Although this can be explained by the fact that the pathogenesis of adenocarcinoma is not related with the bronchus and therefore with basal cells, but with Clara Cells and type-II pneumonocytes, so basal cell hyperplasia and also metaplasia are not required to have reduced protein expression.16
Females with adenocarcinoma were significantly more likely to have methylated MLH1 and MSH2 compared to males. Although, these differences were not observed in squamous cell carcinoma, males were more likely to have hypermethylation of MLH1 and MSH2. These substantial differences have not been consistently noted in the literature,15 at least on the genes under study, perhaps because few studies have been stratified by histological type when assessing hypermethylation differences by gender.15 Since females are more likely to have adenocarcinomas compared to males, differences in hypermethylation frequency due to gender may have been obscured by differences due to histological type.16
Hawes et al. mentioned in their recent work that substantial differences in hypermethylation by gender may suggest the possibility of differential pathways and/or risk factors for NSCLC between genders, with respect to risk factors, especially with regards to the effect of cigarette smoking as well as survival and effectiveness of treatment.17,18 Hormonal factors may account for these differences, although the mechanism that influences the methylation status in NSCLC remains unclear.18
As in our study, Xinarianos et al.19 and Cooper et al.,20 did not find any correlation between reduced MLH1 and MSH2 expression and age, gender, tumour differentiation or TNM stage.
Most of the studies used qualitative methylation-specific PCR in order to detect DNA methylation, a method that can sometimes lead to false positive results and does not distinguish between low and high level methylation. Furthermore, results in these studies have been inconsistent due to varying methylation detection protocols, PCR primers, and study populations, and none have comprehensively studied more than 10 genes.16,17
The variability in results that we observed is probably due to a number of technical factors, such as assay-specific differences including target sites of CpG island loci, primers and the conditions of sodium bisulfite modification. Promoter hypermethylation is one of the mechanisms responsible for the genes silencing,4 although in this work no correlation was found between MLH1 and MSH2 promoter methylation and IHC expression in tumour samples (Tables 6 and 7).
To better understand the role of MMR system in the tumourigenesis of carcinomas of the lung, we conclude that there is a real need for a more robust study covering the differences inherent to tumour histology, heterogeneity and preservation, and finally differences in the study population (age, gender and number of subtyped cases beyond NSCLC), to elucidate other possible mechanisms of altered expression of the hMLH1 and hMSH2, including loss of heterozygosity, chromosomal instability and imbalance mutations in genes of the MMR system,8 and alterations in mRNA transcription.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingFunded by a grant from CIMAGO, Faculty of Medicine, University of Coimbra, Portugal.
Conflicts of interestThe authors have no conflicts of interest to declare.


