


Cystic fibrosis (CF) is a monogenic inherited disease caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein. The disease has a multiorgan involvement, but primarily affects the respiratory and gastrointestinal tracts, and leads to high morbidity and mortality.1 Brazil currently is the sixth highest country worldwide for the number of people diagnosed with CF (PwCF)1 and an incidence of 1 in ∼7,500 newborns.2
Over the last decade, modulator drugs rescuing mutant CFTR traffic and function were developed, thus targeting the fundamental cause of CF.1,3 Recently, a triple combination of two CFTR correctors (elexacaftor and tezacaftor) and a CFTR potentiator (ivacaftor) demonstrated safety and remarkable effectiveness in PwCF carrying the most prevalent F508del mutation.3–5 In phase III trials, the elexacaftor-tezacaftor-ivacaftor (ETI) combination remarkably improved lung function, body mass index (BMI), CF quality of life scores, and sweat chloride concentration (SCC) in either F508del-homozygous or F508del-heterozygous PwCF.4,5 It is estimated that the introduction of ETI into clinical practice will increase the life expectancy of PwCF to >50 years in the United States,6 although it is much lower in countries where ETI is not available.7 This report aimed to assess the real-life efficacy of ETI in Brazilians with CF who earned the right to receive this therapy through court rulings.
After ethics committee approval (CAAE: 15748619.8.1001.0068), written informed consent was obtained from all participants. Twenty-two PwCF were followed by specialized pulmonology physicians at a single CF center in Sao Paulo. All data were documented and processed anonymously, and obtained at baseline, 3 and 6 months after ETI initiation. Lung function (as forced expiratory volume in 1s [FEV1]) was measured according to the recommendations of the American Thoracic Society8 using Brazilian Predictive Values.9 Computed tomography (CT) scan was performed with thin slices (1 mm) in volumetric multi-detector instruments, which allow for analysis of the whole lung parenchyma in high resolution. The SCC was measured by colorimetric method with a reference value for CF as >60 mmol.L–1. The usage of oxygen therapy, frequency of pulmonary exacerbations, BMI, and adverse effects were also included. The quality of life score was assessed using the CF Questionnaire-Revised (CFQ-R).10 For statistical analysis, the Kolmogorov-Smirov test was performed to assess the data normality. Thereafter, paired Student's T-test was used to compare SCC, pulmonary exacerbations, and CFQ-R values, while differences in parameters between F508del-homozygous and F508del-heterozygous cohorts were assessed by unpaired Student's T-test. Finally, General Linear Model for repeated measurements was performed to compare BMI and FEV1 at different time points. A P-value <0.05 was considered significant.
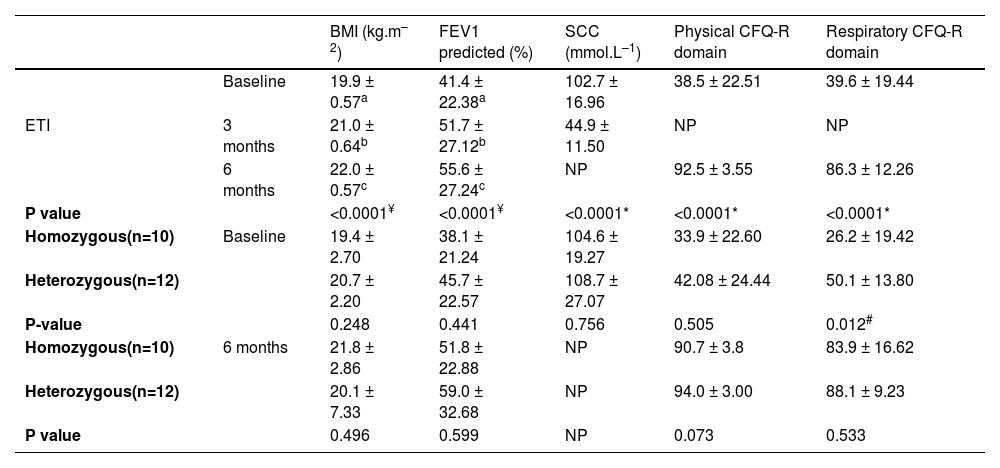
The mean age of the 22 participants was 26.6 (± 9.2) years, all had pancreatic insufficiency and 14 were male (63.6%). All had the F508del mutation on at least one allele, 10 being (45.4%) homozygous for this mutation. Other mutations present in heterozygosis were: 2184delA (1), Q220X (1), A349P (1) G542X (3), R1066H (1), R1066S (1), and R1162X (4). Fifteen individuals (68.2%) had advanced lung disease (FEV1 <40% predicted) and 12 (54.5%) were under continuous home oxygen therapy. The baseline mean of BMI, FEV1, and SCC were 19.9 kg.m–2, 41.4% predicted, and 102.7 mmol.L–1, respectively (Table 1).
Clinical data of 22 participants at baseline, 3 and 6 months after ETI initiation. Data were presented as mean ± standard deviation. Significant results were considered significant when p<0.05.
| BMI (kg.m–2) | FEV1 predicted (%) | SCC (mmol.L–1) | Physical CFQ-R domain | Respiratory CFQ-R domain | ||
|---|---|---|---|---|---|---|
| Baseline | 19.9 ± 0.57a | 41.4 ± 22.38a | 102.7 ± 16.96 | 38.5 ± 22.51 | 39.6 ± 19.44 | |
| ETI | 3 months | 21.0 ± 0.64b | 51.7 ± 27.12b | 44.9 ± 11.50 | NP | NP |
| 6 months | 22.0 ± 0.57c | 55.6 ± 27.24c | NP | 92.5 ± 3.55 | 86.3 ± 12.26 | |
| P value | <0.0001¥ | <0.0001¥ | <0.0001* | <0.0001* | <0.0001* | |
| Homozygous(n=10) | Baseline | 19.4 ± 2.70 | 38.1 ± 21.24 | 104.6 ± 19.27 | 33.9 ± 22.60 | 26.2 ± 19.42 |
| Heterozygous(n=12) | 20.7 ± 2.20 | 45.7 ± 22.57 | 108.7 ± 27.07 | 42.08 ± 24.44 | 50.1 ± 13.80 | |
| P-value | 0.248 | 0.441 | 0.756 | 0.505 | 0.012# | |
| Homozygous(n=10) | 6 months | 21.8 ± 2.86 | 51.8 ± 22.88 | NP | 90.7 ± 3.8 | 83.9 ± 16.62 |
| Heterozygous(n=12) | 20.1 ± 7.33 | 59.0 ± 32.68 | NP | 94.0 ± 3.00 | 88.1 ± 9.23 | |
| P value | 0.496 | 0.599 | NP | 0.073 | 0.533 |
BMI: body mass index; CFQ-R: cystic fibrosis questionnaire-revised; ETI: elexacaftor-tezacaftor-ivacaftor; FEV1: forced expiratory volume in one second; SCC: sweat chloride concentration; NP: not performed.
After 3 and 6 months of ETI initiation, the mean improvement of FEV1 was 10.3% and 14.2% points (Table 1). CT scans revealed a marked improvement in lung abnormalities, although bronchiectasis persisted (Fig. 1). Only one individual remained under continuous home oxygen therapy after ETI therapy, representing a 91.6% reduction in supplementary oxygen necessity. The annualized number of pulmonary exacerbations per individual after ETI initiation (mean 0.22 ± 0.42) was significantly lower (p<0.0001) compared to the frequency in the year before ETI initiation (mean 2.96 ± 1.77).
. (A) At baseline, diffuse bronchial thickening, intrabronchial mucus plugging, and bilateral bronchiectasis in lower lobes were evidenced. (B) After 6 months of ETI therapy, there was a significant reduction in mucus plugging and bronchial thickening.")
Chest CT scan of a 22-year-old female with CF (F508del/F508del genotype). (A) At baseline, diffuse bronchial thickening, intrabronchial mucus plugging, and bilateral bronchiectasis in lower lobes were evidenced. (B) After 6 months of ETI therapy, there was a significant reduction in mucus plugging and bronchial thickening.
Triple therapy improved BMI with a mean increase of 1.1 and 2.1 kg.m–2 in 3 and 6 months, respectively, from baseline. Similar findings were observed for the mean of SCC, which dropped 49.7 mmol.L–1 after 3 months of ETI, indicating the rescue of CFTR function. Regarding the quality of life questionnaire (CFQ-R), the mean values increased significantly in both physical activity (+54 points) and respiratory (+46.7 points) domains after 6 months of ETI.
Overall, ETI therapy was well tolerated and 13 individuals (59.1%) demonstrated no adverse effects. There was no ETI discontinuation and adverse effects observed in the remaining individuals were classified as mild or moderate and included: rash (1), diarrhea (2), abdominal pain (2), and altered values in liver enzymes (CPK and ALT) (4).
This is the first real-life study in Brazil to demonstrate an association of ETI therapy with the significant improvement of predicted FEV1, BMI, and quality of life scores in PwCF in Brazil. Triple therapy also reduced SCC, the need for oxygen therapy, and the frequency of pulmonary exacerbations in a 6-month follow-up. Furthermore, ETI therapy was safe and well tolerated, which aligns with other clinical studies in PwCF carrying at least one F508del-CFTR that demonstrated a rapid clinical improvement after ETI initiation3,11 and a low discontinuation rate (<2%).4,5
Although F508del-homozygous participants exhibited the worst CFQ-R scores in the respiratory domain at baseline, similar clinical effectiveness was observed in the F508del-homozygous and F508del-heterozygous cohorts after ETI therapy (Table 1). Additionally, the improvements in lung function and structure, and nutritional status were sustained for up to 6 months in both cohorts. Similar results were found in a post-approval trial, although the mean of FEV1 increased 9.76% points after 6 months of ETI,12 compared to 14.2% points increase in this study. Lung function changes may have been larger in our report since PwCF were naïve to modulators, while half of the participants in the former study were already using other modulators before switching to ETI.12
Pulmonary exacerbations are deleterious events that frequently lead to accelerated decline in lung function, hospitalization, and worse quality of life.4,5 Following lung function improvement in this study, a significant reduction of pulmonary exacerbation occurrence was also associated with the usage of ETI therapy, which may have contributed to the decreased respiratory symptoms and increased self-perceived quality of life.
In summary, this report demonstrated the safety and efficacy of ETI in PwCF in a real-life setting in Brazil. Despite the small sample size, ETI had remarkably positive effects on all clinical outcomes evaluated in the 6 months follow-up, highlighting its impact not only on symptoms and complications but also on individuals’ physical and social well-being. Therefore, the introduction of ETI into clinical practice in Brazil may represent a life-changing therapy for many PwCF.


