


Cystic fibrosis (CF) is a multisystemic disease with clinical heterogeneity, and progressive lung disease remains the major cause of morbidity and mortality.1 Some CF patients can be diagnosed later in life. Studies have suggested that patients with a delayed diagnosis survive better, reflecting the prevalence of mutations with a less severe phenotype.2 Some rarer mutations have been associated with mild CF and delayed diagnosis; one such example is the p.Val232Asp mutation.3
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that frequently spreads to involve most muscles leading to severe disability.4,5 Progressive weakness of respiratory muscles leads typically to respiratory failure, and lung function tests are valuable in predicting its course. The decline in pulmonary function closely correlates with death for patients not managed with respiratory aids (non-invasive ventilatory support – NVS, and cough assistance techniques).6
A twenty-two years old male, with a history of allergic asthma and rhinitis since childhood, with no current follow-up. No smoking or relevant family history. He presented with weakness of upper right limb, and muscle cramps, for four months. In association, he referred to worsening of cough and sputum. The neurologic examination revealed atrophy of the right forearm and arm, hyperreflexia and weakness of the upper limbs. Besides the presence of fever, the physical examination was normal. The patient was admitted to the Neurology department for clinical evaluation. Laboratory studies showed a slight elevation of infection markers. Arterial blood gases test were normal. Brain/vertebral magnetic resonance imaging and electromyography (EMG) were normal. Chest computed tomography scan showed cylindrical bronchiectasis with wall thickening, with a tree-in-bud micronodular pattern (Fig. 1, panel A–C). Due to recurrent respiratory infections since childhood, with cough and chronic bronchorrhea and, isolation of methicillin-susceptible Staphylococcus aureus in bronchial secretions, CF was suspected. The sweat test was 91mmol/L, and the genetic study identified p.Val232Asp and p.Phe508del mutations. Both neurological and respiratory symptoms improved with antibiotic therapy, so the patient was discharged. He began follow-up and treatment at the CF outpatient clinic, and radiological improvement was noticeable (Fig. 1 – panel D). No other organ involvement was observed. Spirometry revealed moderate obstructive ventilatory pattern (FVC: 2690mL/62.9%; FEV1: 2440mL/65.9%). For the following three months, he experienced worsening of the neurological deficits, with severe weakness of the four limbs (upper right limb with muscle atrophy, Fig. 1 – panel E–F), with an abnormal gait. Repetition of EMG showed neurogenic injury and active denervation, and with the clinical background were indicative of a definitive ALS diagnosis. At this point, he was experiencing a decline in respiratory muscle function, with a VC of 2100mL, decreasing 15% in the supine position, a peak cough flow of 270L/min and elevation of partial carbon dioxide pressure (47.8mmHg). Home mechanical ventilation was initiated with NVS (assisted/controlled mode, with oronasal mask) during sleep, and cough assistance techniques were optimized. Although he had not reported orthopnea or other sleep related symptoms, after beginning NVS he reported better sleep quality without orthopnea and a global improvement during the day; symptoms that he previously undervalued. Palliative care team was also involved.
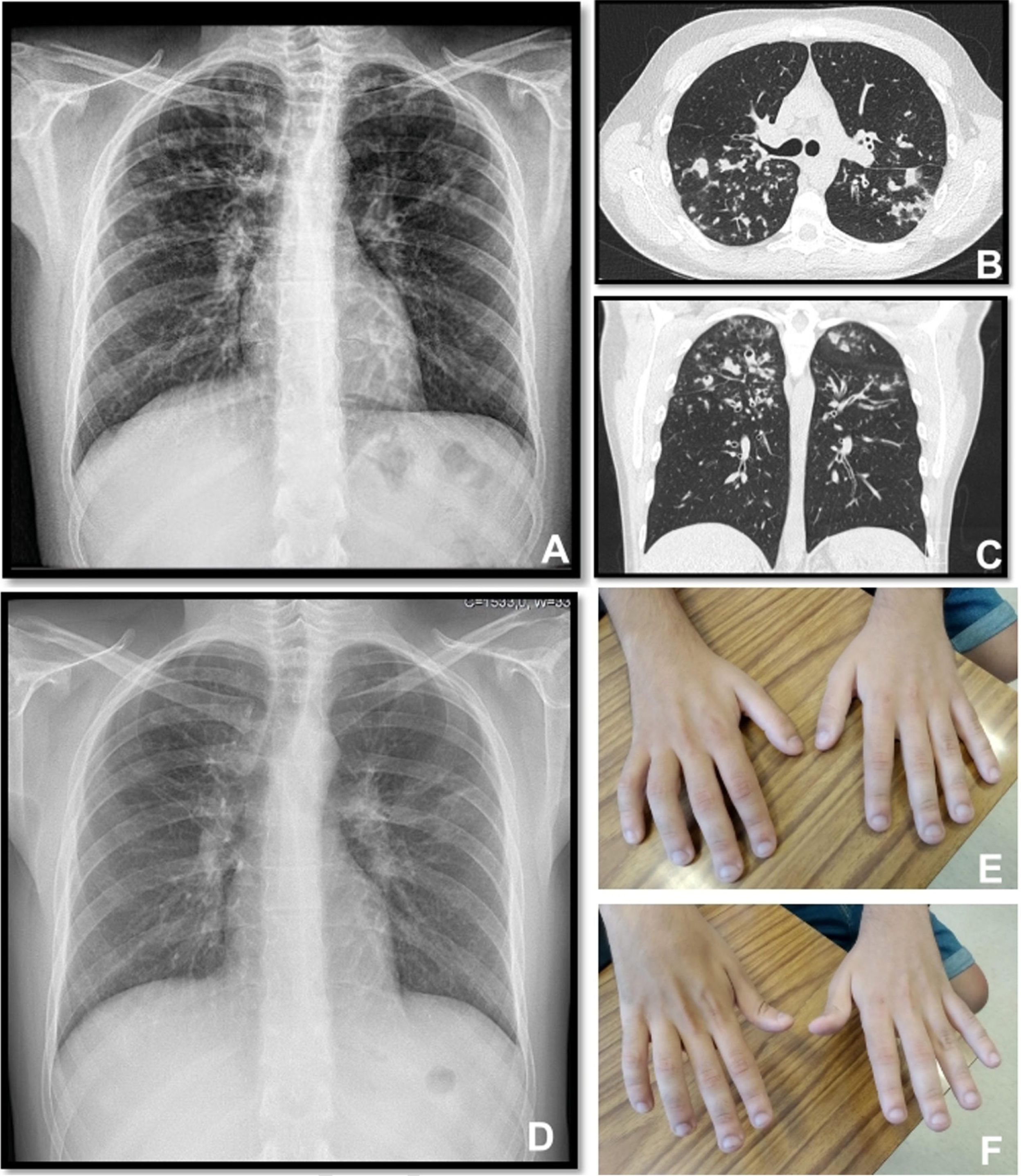
Panel A, B and C: Chest x-ray and thoracic computed tomography showed cylindrical bronchiectasis with wall thickening associated with a tree-in-bud micronodular pattern; Panel D: Chest x-ray with radiological improvement, after CF therapy was initiated; Panel E-F: severe weakness of upper limbs (worse in the upper right limb with muscle atrophy).
The authors present a challenging clinical case. On the one hand, we intend to show an example of a late diagnosis of CF and a young-onset of ALS. Given the diversity and versatility of CF presentation, the diagnosis may go unnoticed for years, therefore it is essential to maintain high suspicion regarding the association of certain clinical characteristics. Moreover, this case exemplifies a young-onset of ALS; while it has a progressive course, young patient's survival is frequently longer and the percentage of bulbar onset is lower in the young-adult type. Also, this patient had no family history of ALS. On the other hand, the simultaneous diagnosis of CF and ALS presents several challenges in follow-up and treatment. Airway clearance techniques, a key feature in treating CF, will be compromised due to the inevitable respiratory muscle weakness and ineffective cough associated with ALS with progressive increase of ventilatory dependence. Patients with strictly non-bulbar presentation (as in this case) retain effective speech and swallowing although their VCs can decrease to <100mL, and all breathing tolerance can be lost and substituted by effective continuous NVS. This case also unveils the psychological issues arising when treating a young patient with two progressive and incurable diseases. The personal impact of the functional decline, the increasing assistance over time and lifestyle changes resulting from such a complicated situation will be hard to manage, highlighting the importance of a multidisciplinary team.
To the best of our knowledge this is the second case reported with CF and ALS.7 Previously reported was a case of a 30-year-old female, diagnosed with CF 16 years earlier. Genotyping revealed a complex heterozygote of p.Gly542 and p.Pro67Leu mutations. The authors suspected that TDP-43 dysfunction could be linked to both pathologies, but the patient did not display TDP-43 mediated aberrant splicing of the CFTR gene. Alternative reasons were also considered.
In the last few years, the treatment of CF has experienced a period of evolution, with survival estimated around 40 years-old.1 Survival in ALS is slightly longer in young patients, and improved due to the use of NVS; despite this, the average survival after the diagnosis is up to 2–4 years.6 Therefore, the authors realize that concurrent CF and ALS might have poor prognosis.
Conflicts of interestThe authors have no conflicts of interest to declare.


