




Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease that up to now has been associated with a poor prognosis. However, the results of the INPULSIS and ASCEND trials and the approval of nintedanib and pirfenidone have marked the beginning of a new era for IPF patients. Questions remain, however. Should these drugs be used earlier? What effect will they have on more severe disease? Will their effects last beyond the trial period? This manuscript is the outcome of a multidisciplinary meeting between pulmonology, radiology, and pathology clinicians on the use of antifibrotic agents in IPF. In our opinion, the existing data show that pirfenidone and nintedanib slow functional decline in early stages of disease. These drugs also appear to result in therapeutic benefits when administered to patients with advanced disease at diagnosis and maintain effective over time. The data also suggest that continuing antifibrotic therapy after disease progression may confer benefits, but more evidence is needed. Early diagnosis and treatment are crucial for reducing functional decline, slowing disease progression, and improving quality of life.
Idiopathic pulmonary fibrosis (IPF) is a progressive and deadly disease with a mean survival of 3–5 years from the time of diagnosis. It is more common in men, smokers, and people over 50 years old.1,2 The condition is characterized by dyspnea on exertion, non-productive cough, crackles on auscultation, and digital clubbing. It is diagnosed using a specific combination of radiologic and/or histopathological patterns of usual interstitial pneumonia (UIP) and exclusion of other known causes of UIP.1,3 General physicians must be able to recognize the symptoms of IPF to ensure the timely referral of suspected cases, and it is essential that these are then evaluated by a multidisciplinary team with experience in interstitial lung diseases to ensure an accurate diagnosis.1,4–8
This manuscript is the outcome of a multidisciplinary meeting in which pulmonology, radiology, and pathology clinicians came together to debate topics related to the use of antifibrotic drugs in the treatment of IPF.
IPF treatment: a recent (r)evolutionIPF was initially considered to be a chronic inflammatory process and therefore early treatment strategies aimed to eliminate or suppress the inflammatory component. In 2000, the first international guidelines on IPF diagnosis and treatment recommended corticosteroids and immunosuppressive/cytotoxic agents (azathioprine or cyclophosphamide) as “standard treatment”, despite the weak evidence available.9 This targeting of inflammatory pathways, however, produced disappointing results.4,10–12
Understanding of IPF pathobiology improved significantly following the publication of the 2000 IPF guidelines and it was proposed that the disease might be the result of an aberrant healing response to recurrent alveolar epithelial cell injury.13,14 Guidelines published in 2011 concluded there was insufficient evidence to continue to recommend corticosteroid and immunomodulatory agents as standard therapy for IPF.15 The results of the IFIGENIA study, a double-blind clinical trial designed to investigate the potential role of antioxidant pathways in IPF, were published in 2005. The trial, which compared prednisolone plus azathioprine with prednisolone plus azathioprine plus N-acetylcysteine (NAC), showed a significantly reduced decline in forced vital capacity (FVC) (p=0.02) and diffusion capacity of the lung for carbon monoxide (DLCO) (p=0.003) in patients treated with NAC combined with prednisolone and azathioprine after 52 weeks.16 Although the trial had some methodological pitfalls, this triple therapy became the new standard treatment for IPF.
Around the same time, pirfenidone, an orally bioavailable synthetic compound with antifibrotic, anti-inflammatory, and antioxidant effects, was being studied in clinical trials in Japan.17,18 Although the exact mechanism of action of pirfenidone in IPF is not yet completely understood, its anti-inflammatory effects are believed to result from the suppression of tumor necrosis factor α (TNF-α), interleukin (IL)-6, IL-12, and IL-8,19 while its anti-fibrotic effect is thought to result primarily from inhibition of expression of transforming growth factor beta – a profibrotic cytokine – although other pathways have been suggested.20 The first Japanese trial, a phase II, multicenter, randomized, double-blind trial, showed no significant treatment effect for pirfenidone on the primary endpoint, which was a change from baseline in the lowest oxygen saturation by pulse oximetry [SpO2] during a 6-min exercise test. It did, however, show that the high-dose treatment (1800mg/day) had a positive effect in reducing VC decline, a secondary endpoint, at 9 months.17 In addition, acute exacerbations were only observed in the placebo group during the same period. A subsequent multicenter, randomized, double-blind phase III clinical trial of 275 IPF patients randomly assigned to pirfenidone 1800mg/day, pirfenidone 1200mg/day, or placebo for 52 weeks, showed a significantly slower decline in FVC and better progression-free survival in the two pirfenidone groups compared with placebo.18 Based on the results of these two studies, pirfenidone was approved for IPF treatment by the Japanese authorities in 2008.
Meanwhile, pirfenidone was also being studied in Europe and in the United States of America (USA) in the CAPACITY trials.21 The placebo-controlled phase III trials, CAPACITY 004 and 006, showed contradictory results, and the primary endpoint of a significant reduction in FVC decline at week 72 was only met in study 004. However, the pooled analysis revealed a significant reduction in the mean decline of absolute and percent predicted FVC and the number of patients with an FVC decline ≥10% at week 72 in the 2403mg/day pirfenidone group.21 In relation to the secondary endpoints, the patients in the 2403mg/day group also had longer progression-free survival and a lower rate of decline in mean 6-min walk distance (6MWD) than patients in the placebo group.21 The results of this pooled analysis led to the approval of pirfenidone for IPF treatment by the European Medicines Agency (EMA) in 2011, although they were not sufficient to convince the US Food and Drug Administration (FDA).
Despite the approval of pirfenidone in Japan and Europe, the triple therapy persisted as the treatment regimen of choice in many countries until the results of the PANTHER study were published.11 The PANTHER study was a randomized clinical trial with three arms: a triple therapy arm, an NAC arm, and a placebo arm. The triple therapy arm was stopped after a mean follow-up of 32 weeks because of higher mortality and hospitalization rates than in the other two groups.11
Despite some initial controversy, this event marked the end of triple therapy. Moreover, on completion of the PANTHER study in 2014, no significant differences were detected between NAC and placebo for changes in FVC (primary endpoint) or for death rates or frequency of acute exacerbations (secondary endpoints).22 Recent publications describing a therapeutic effect for NAC in a subgroup of IPF patients expressing the TOLLIP gene polymorphism rs375092023 and a potential benefit of inhaled acetylcysteine,24 have rekindled the discussion about the potential therapeutic role of NAC and antioxidants as a therapeutic group in IPF.
After the evaluation of the CAPACITY trials,19 the ASCEND trial, which included 555 patients from the USA, Europe, and Australia randomly assigned to receive 2403mg/day of pirfenidone or placebo for 52 weeks showed that the treatment group had a significantly reduced absolute FVC decline, fewer patients with an FVC decline ≥10%, better progression-free survival (defined as the time to occurrence of a confirmed absolute decrease of ≥10 percentage points in percent predicted FVC, a confirmed decrease of ≥50m in the 6MWD, or death), and fewer patients with a decrease of ≥50m in the 6MWD.25 Moreover, pooled data from the CAPACITY and ASCEND trials evidenced a reduction in both all-cause and IPF-related mortality after one year.25 These results further supported the efficacy of pirfenidone in IPF and led to FDA approval in 2014.
In the same year, nintedanib also received FDA approval after the INPULSIS trials.26 Nintedanib is a small molecule tyrosine kinase inhibitor that targets the receptors of platelet-derived growth factor, fibroblast growth factor, and vascular endothelial growth factor. Blockage of these receptors may inhibit downstream signaling cascades of fibroblasts and myofibroblasts, attenuating the development of aberrant fibrosis.27
In the phase II nintedanib trial TOMORROW, which included 432 IPF patients randomized to one of four doses of nintedanib (50mg od, 50mg bid, 100mg bid, and 150mg bid) or placebo, a significant reduction in FVC decline after 52 weeks was seen in the 150mg bid group compared with placebo.28 This subgroup of patients also had significantly fewer acute exacerbations and a smaller mean decrease in the St. George's Respiratory Questionnaire score. Two subsequent large phase III trials, INPULSIS 1 and 2, comparing 150mg bid of nintedanib with placebo in 1066 patients, showed a reduction in the annual rate of decline in absolute and percent predicted FVC (primary endpoint) and an increase in time to the first acute exacerbation for nintedanib.26 With these results, the EMA few months after the FDA approved nintedanib for the treatment of IPF in January 2015.
A new era emerged for patients with IPF following the INPULSIS26 and ASCEND25 trials and the approval of nintedanib and pirfenidone.29 The two drugs were identified in national and international guidelines on the diagnosis and treatment of IPF as being efficacious in slowing functional decline and disease progression in IPF patients.1,30–33
The nintedanib and pirfenidone trials showed similar efficacy results but raised logical questions related to how the drugs would perform in the real world, outside the restrictions of the trials. The inclusion criteria varied somewhat between the INPULSIS (nintedanib) and ASCEND (pirfenidone) trials. The pirfenidone trials21,25 included patients with percent predicted FVC 50–90%, predicted DLCO 30–90% and high-resolution computed tomography (HRCT) images indicating either definite or possible UIP, however with confirmation by surgical lung biopsy. The nintedanib trials,26,28 by contrast, included patients with percent predicted FVC >50%, predicted DLCO 30–79%, and HRCT images indicating definite or possible UIP. Following publication of the trials, several post-marketing surveillance studies and subgroup analyses investigating effects according to age or stage of disease were undertaken in order to answer several questions: If these drugs are efficacious in slowing the progression of IPF, should they be used earlier? How will they behave in patients with more severe disease? Will their effects extend beyond the trial period?29,34
When should IPF treatment be started?The most controversial question currently surrounding IPF treatment is probably when it should be started. We know that IPF is a progressive disease and that both pirfenidone and nintedanib slow functional decline. However, some patients remain stable for several months and it has been argued that in a stable patient with mild disease, treatment should be postponed until functional decline begins. Nevertheless, it is impossible to predict the rate or severity of disease progression in each patient or to know when they will experience an acute exacerbation. Moreover, we cannot be sure that deleterious subclinical changes are not taking place in patients without signs of significant functional deterioration.
In a recent post hoc subgroup analysis of pooled data from the INPULSIS trials, Kolb et al. described similar rates of FVC decline in nintedanib-treated patients with percent predicted FVC >90% and ≤90% (Fig. 1).35 Other pre-specified subgroup analyses have shown similar results, with a consistent effect observed for nintedanib in patients with FVC ≤70% and >70%.36
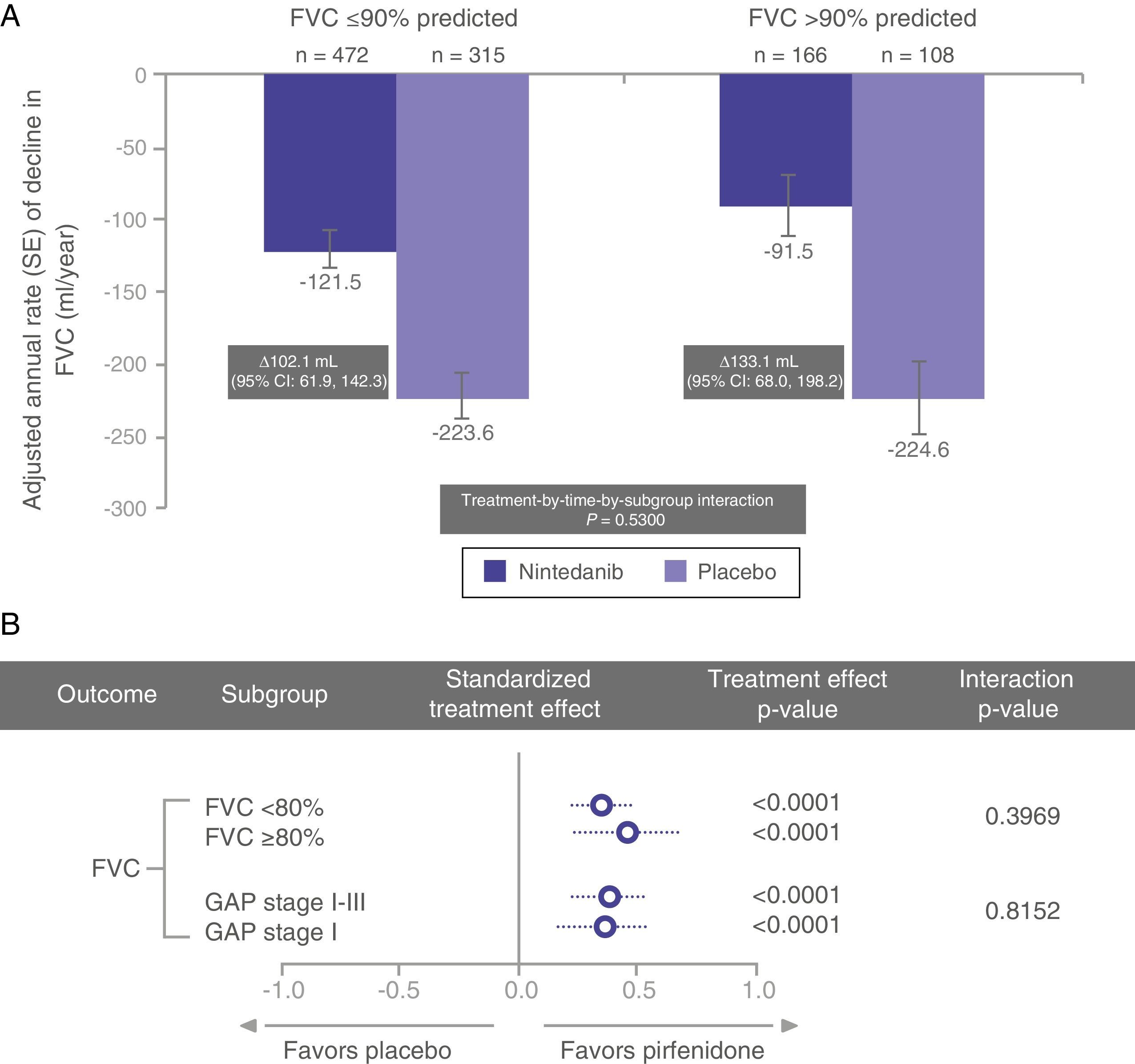
In a post hoc analysis of pooled data from the CAPACITY and ASCEND trials, Albera et al.37 showed similar clinically significant disease progression (decline in FVC, 6MWD, and dyspnea measured by the University of California, San Diego Shortness of Breath Questionnaire) at 12 months in pirfenidone-treated patients with more preserved lung function (FVC ≥80% or GAP stage I) and less preserved lung function (FVC <80% or GAP stage II–III) at baseline. The GAP index is a multidimensional IPF staging model that takes into account gender (G), age (A), and 2 lung physiology variables (P) (FVC and DLCO); it distinguishes between stages I, II, and III.38 The magnitude of the pirfenidone treatment effect was comparable between the subgroups, regardless of whether lung function was classified as using FVC or the GAP model (Fig. 1).37
The above data suggest that patients diagnosed in early stages of disease experience significant functional deterioration and that both nintedanib and pirfenidone have a positive effect in terms of slowing progression in these early stages (Fig. 2), clearly supporting the recommendation to initiate treatment immediately after IPF diagnosis and highlighting the importance of prompt diagnosis.
Severe disease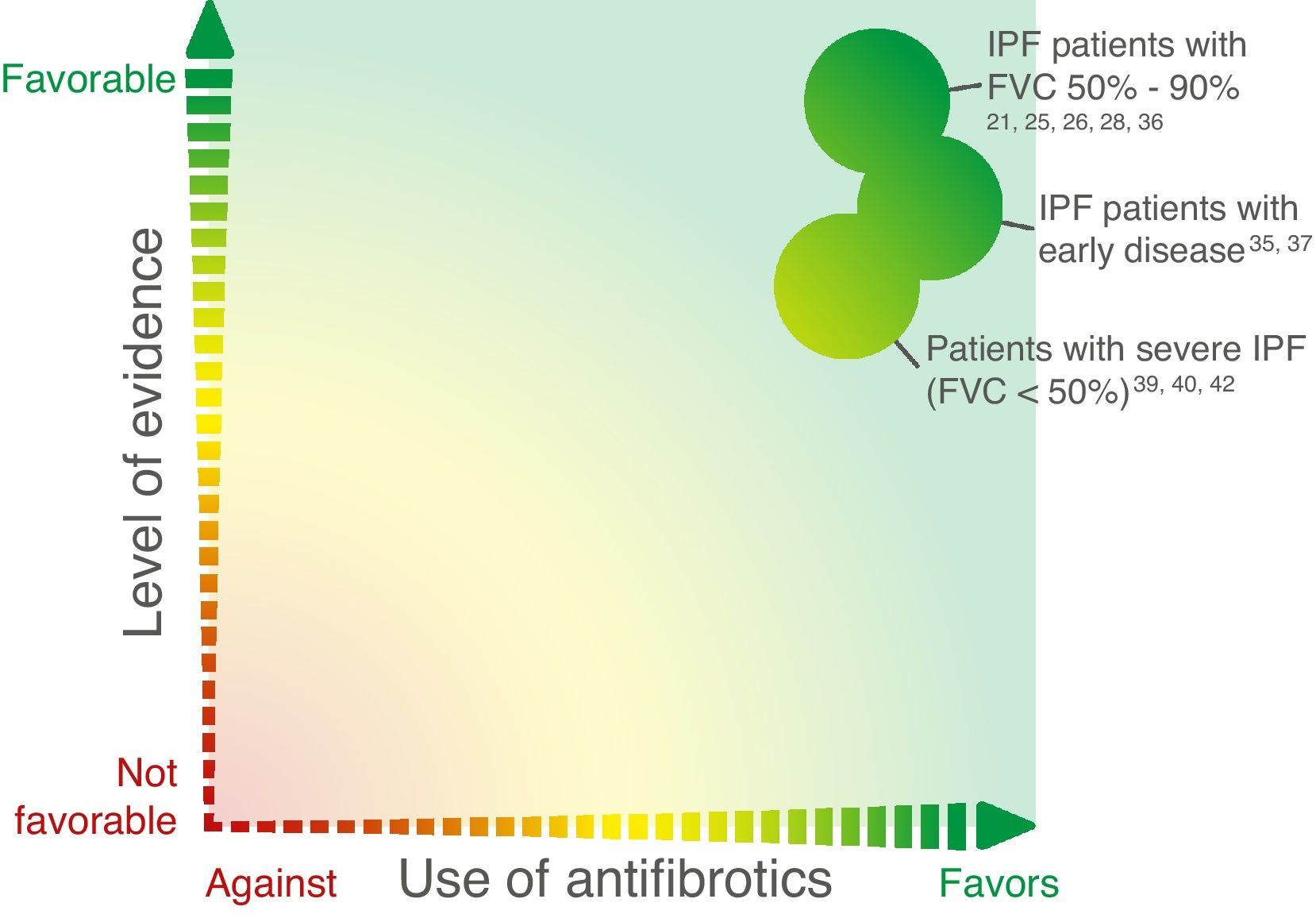
Percent predicted FVC <50% is usually associated with severe disease. As stated earlier, this was an exclusion criterion in both the pirfenidone and nintedanib trials.21,25,26,28 Information about the potential effects of antifibrotic agents in patients with severe disease is thus scarce, and overall, the treatment indications outlined by the health authorities exclude patients with this grade of functional impairment.
Some interesting data in this respect, however, have emerged from post-marketing surveillance studies. In the open-label INPULSIS-ON extension trial, patients who started treatment with nintedanib 150mg bid with percent predicted FVC ≤50% (n=24) showed a similar absolute mean change in FVC from baseline to week 48 as those with FVC >50% at baseline (−62.3mL vs. −87.9mL),39 suggesting that nintedanib may have a similar therapeutic effect in advanced forms of the disease.
In a real-life retrospective study of pirfenidone in several Italian interstitial lung disease centers, Harari et al.40 evaluated 128 IPF patients stratified according to FVC and GAP stage, and reported a greater reduction in decline in percent predicted FVC in patients with moderate to severe disease.
In Japan, a retrospective evaluation of 1371 patients who began pirfenidone in the first year after its approval for IPF treatment detected functional and symptomatic stabilization, regardless of degree of functional impairment.41 Two-thirds of the patients were considered to have advanced disease (stage III–IV, according to a four-stage Japanese IPF staging model based on partial pressure of arterial oxygen at rest and SpO2 during the 6MW test).41
At the recent 2016 European Respiratory Congress, Costabel and colleagues42 reported on the results of a comparison of 54 patients with percent predicted FVC ≤50% with 530 patients with FVC >50% in the RECAP trial, which was an extension of the CAPACITY trials. Although a higher treatment discontinuation rate was observed in the subgroup with more advanced disease, long-term treatment with pirfenidone resulted in a similar rate of FVC decline in both groups.
Despite the relatively small number of patients, the above data suggest that both nintedanib and pirfenidone have a positive effect in patients who have advanced disease at diagnosis (Fig. 2).
Is the treatment effect maintained after 52 weeks?Another question that post-marketing surveillance studies and extension trials such as RECAP43 and INPULSIS-ON44,45 have attempted to answer is whether or not the positive effect observed for nintedanib and pirfenidone in terms of slowing FVC decline is maintained after 52 weeks.
In 2014, Valeyre et al.46 published the results of a long-term safety assessment of patients treated with pirfenidone in the CAPACITY trials. Although safety was the primary focus of the study, the authors demonstrated that treatment with pirfenidone for up to 7.7 years (median, 2.6 years; range, 1 week–7.7 years) was not only well tolerated but also showed a persistent positive therapeutic effect.46
More recently, in an oral communication at the 2016 ERS Congress (London, 3–9 September 2016), Noble et al.47 presented data from the RECAP extension study on the long-term effects of pirfenidone on percent predicted FVC and showed that the drug maintained its effect over more than 3 years.
Finally, in a recent presentation of results from the INPULSIS-ON extension study, Crestani showed that FVC decline was similar to that observed in the original INPULSIS trials, suggesting that like pirfenidone, nintedanib also maintained its effect for over 3 years.44,45
What should be done once disease progression is confirmed?Disease progression in IPF is defined as a sustained decline of more than 10% in FVC and/or of 15% in DLCO. Lung function is usually evaluated every 6 months following initiation of treatment. Once disease progression has been established, the treating physician(s) can choose to suspend the drug and administer palliative care only; suspend the drug and switch to an alternative therapy; add an alternative therapy; or continue the drug despite the functional decline. There is insufficient evidence, however, to support any of these options. In May 2016, Nathan et al.48 published the results of a pooled analysis of patients from the CAPACITY and ASCEND trials showing that in the subgroup of patients who experienced a functional decline in FVC >10% after 6 months, those who continued to receive pirfenidone had a lower risk of subsequent FVC decline or death. These results suggest that maintaining antifibrotic therapy may confer benefits even after disease progression is confirmed.
Bonella et al., 49 in a German multicenter observational study of nintedanib in IPF, reported that a subgroup of patients who had shown disease progression under pirfenidone, achieved clinical and functional stability when switched to nintedanib. A combination regimen of pirfenidone and nintedanib has also been proposed, as theoretically it would allow synergistic activity in different fibrotic pathways. One safety and pharmacokinetics study reported acceptable tolerability for nintedanib administered alone or with pirfenidone and also showed that nintedanib bioavailability may decrease with the co-administration of pirfenidone.50 Despite these findings, more data and studies are needed to guide treatment strategies in patients with disease progression.
What is the impact of adverse events induced by nintedanib or pirfenidone?The most common adverse events reported for nintedanib in the TOMORROW28 and INPULSIS trials26 were diarrhea, nausea, and vomiting, which were more prevalent in the 150mg dose group. Although 60% of patients in the INPULSIS trials developed diarrhea, only 4% discontinued treatment for this reason. In a pooled analysis from the TOMORROW and INPULSIS trials, Richeldi et al.51 found that 20.6% of patients in the nintedanib group prematurely discontinued treatment due to overall adverse events versus 15.0% in the placebo group. Nearly 30% of patients in both groups experienced one or more serious adverse events. Adverse events due to nintedanib, however, are usually mild or moderate and can be easily managed in most patients.
An interim safety analysis of the INPULSIS-ON extension study44,45 showed that patients who completed 96 weeks of nintedanib treatment experienced fewer adverse events (316.8 events per 100 patient-years) than those who completed the initial 52-week evaluation (632.6 events) and those who initiated treatment after 52 weeks on placebo (550.6 events). A similar situation was observed for serious adverse events (33.9 vs. 36.7 and 38.3 events per 100 patient-years) and events leading to treatment discontinuation (14.6 vs. 21.4 events per 100 patient-years in the other two groups). The author concluded that these interim results from the INPULSIS-ON trial support the long-term efficacy and safety of nintedanib in patients with IPF.
The most common adverse events during the pirfenidone CAPACITY19 and ASCEND trials25 were gastrointestinal events (nausea, dyspepsia, vomiting, and anorexia) and skin disorders (rash, photosensitivity). The events were generally mild or moderate, reversible with dose reductions, and without clinically significant consequences.
In a comprehensive analysis of safety across four clinical trials evaluating pirfenidone in patients with IPF, the more frequent adverse events reported were also mild to moderate nausea (40%), dyspepsia (21%), vomiting (18%), and rash (26%). These events rarely led to treatment discontinuation, despite the longer median duration of exposure to pirfenidone (2.6 years).46 This study also showed that while gastrointestinal and skin-related adverse events are relatively common at the beginning of treatment, they decrease considerably after the first 6 months and remain low during the course of treatment.
Ogura et al., 41 in a prospective post-marketing surveillance study of patients with IPF administered pirfenidone in the first year after its launch in Japan, found a similar safety profile to that reported in the Japanese phase II and III trials, with 64.6% of patients experiencing at least one adverse event. The most common events were decreased appetite, photosensitivity, and nausea, and these resolved or improved in the majority of cases.
The results of the RECAP open-label extension study of pirfenidone showed that the type and frequency of adverse events after 60 weeks of treatment were similar to those observed in the initial 72-week phase III CAPACITY trials and rarely resulted in early discontinuation of therapy.43 The incidence of nausea and photosensitivity was 35.4% and 11.9% in the CAPACITY trials versus 32.0% and 11.8% in the RECAP study. Rash and diarrhea, by contrast, were less common in RECAP (18.0% and 16.9% vs. 31.0% and 27.0%, respectively).21,43
ConclusionsBased on the available data from clinical trials and extension studies, we can conclude that both pirfenidone and nintedanib have a significant effect on FVC decline, regardless of disease stage assessed by FVC or GAP. Moreover, the RECAP and INPULSIS-ON extension trials showed that the effect was maintained over time. Both nintedanib and pirfenidone were found to prolong survival, with a reduction in both IPF-related and all-cause mortality.
Despite the relatively high rates of adverse events, which are mostly gastrointestinal in nature, the existing data support the recommendation that treatment with nintedanib or pirfenidone should be considered when IPF is diagnosed, regardless of disease stage. Since IPF is a progressive lung disease, early diagnosis and treatment are crucial for slowing functional decline, reducing symptoms, and improving quality of life.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of interestThe authors have no conflicts of interest to declare.


