



Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic syndrome caused by mutations in the NF1 gene that encodes neurofibromin protein which acts as a tumor suppressor. It has a wide range of clinical features, it is characterized by cutaneous signs, notably neurofibromas, café-au-lait macules, iris hamartomas as well as axillary and inguinal freckling. It affects one in every 3000 people, 30–50% of which are sporadic form cases.1
Respiratory involvement may be result from diverse ailments such as pulmonary hypertension, kyphoscoliosis, bilateral diaphragmatic paralysis, etc., as well as sporadic cases of diffuse pulmonary disease associated with NF1 (NF1-DLD).2 We present a patient with both concomitant diseases.
A 62-year-old woman with a past smoking history of 100packs/year without preceding pulmonary pathology but previously diagnosed with NF1 (sporadic form) in her youth. She had been diagnosed a year earlier of an infiltrating ductal carcinoma in her right breast treated with sequential neoadjuvant therapy and subsequent mastectomy. She was treated in Emergency Department because of fever and cough in the previous 4 days. Physical examination showed bilateral wheezing without other relevant findings, except for countless neurofibromas (Fig. 1A). Leukocytosis with left shift and acute respiratory failure was observed. Chest radiograph displayed thickening of peribroncovascular interstice with left basal predominance (Fig. 1B, C) and multiple and bilateral irregular thin-walled aerial cysts were seen in the high resolution computed tomography (CT), converging in upper zones mimicking emphysema (Fig. 1D, E). Her pulmonary function tests were: FVC 1400cc (61.7%); FEV1 890cc (47%); FEV1/FVC 63.4 (77.3); DLCOsb 29.2%; DLCO/VA 39.3%. Based on these findings, diagnosis of NF1-DLD was established (without any other organs involved). Following oxygen, bronchodilators, antibiotics (levofloxacin) and corticosteroids treatment, she presented a favorable outcome and was discharged from hospital after 4 days.
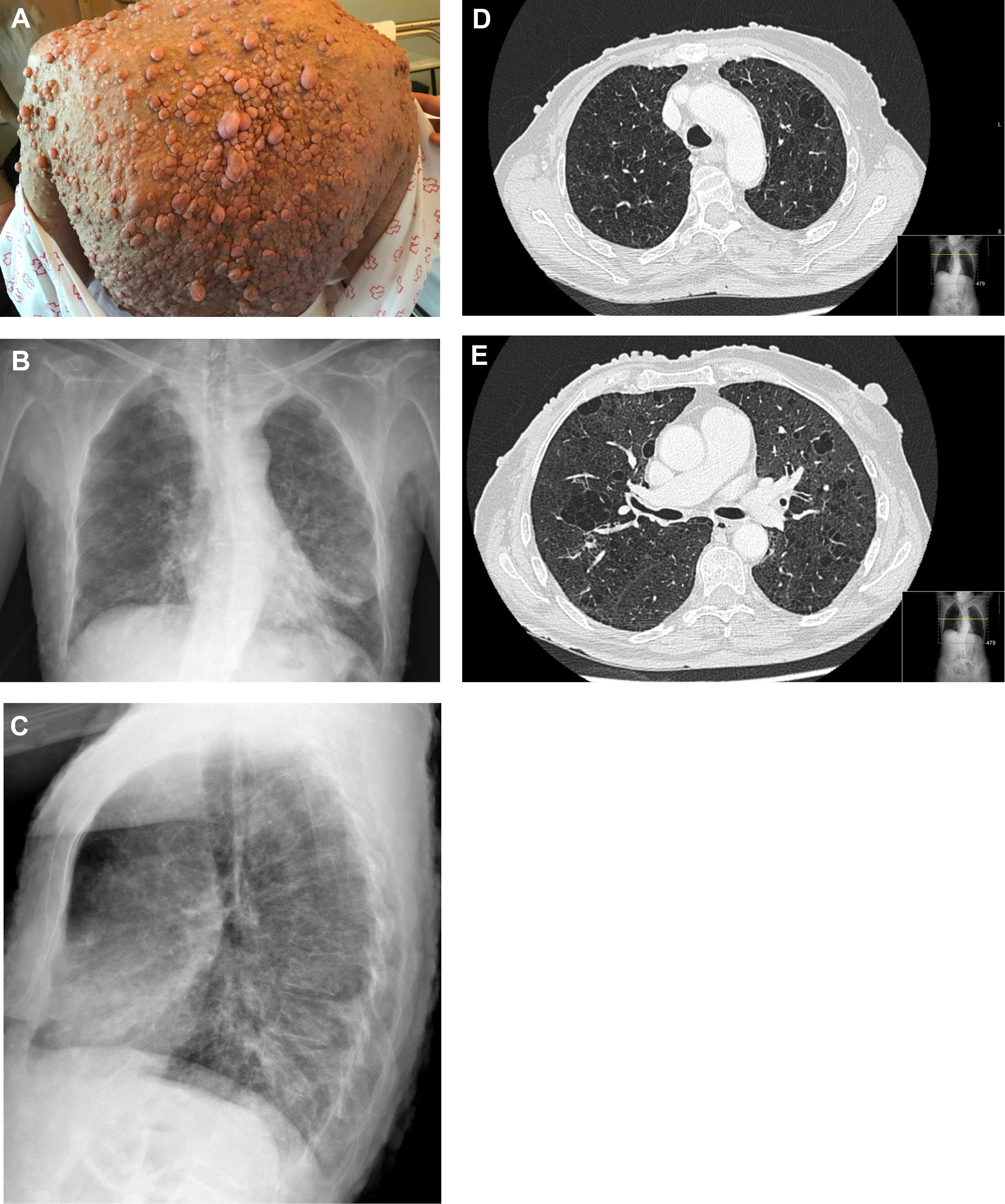
(A) Multiple cutaneous neurofibromas in a NF1 patient. (B, C) Chest X-ray in which thickening of the peribroncovascular interstice with left basal predominance is observed. (D, E) High resolution computed tomography (axial plane and pulmonary window) through upper lobes above and below carina showing multiple thin-walled irregular aerial cysts, that tend to converge in upper areas, with similar appearance to emphysema.
Three main Neurofibromatosis types have been described: 1 (NF1), 2 (NF2) and schwannomatosis. NF1 is the most frequent subgroup, characteristics of which are described above.1 NF2 is a hereditary syndrome, caused by mutations in NF2 gene that encodes merlin protein, a tumor suppressor, which occurs in 1 out of 25,000 inhabitants, without lungs being involved. Schwannomatosis, on the other hand, is caused by mutations in SMARCB1 and LZTR3 tumor suppressor genes and affects 0.58 in 1,000,000 people. Familial and sporadic cases have also been described.
NF1-DLD was first described by Davies4 and has an estimated prevalence of 7–23% of NF1 patients. Respiratory symptoms are often weak and pulmonary function tests can show any pattern type, including obstructive (43%), restrictive (37%) or mixed (17%). Carbon monoxide diffusing capacity is usually affected at an early stage. Common chest CT findings include upper lobe predominant cystic and thin-walled bullous disease (25%), basilar reticular abnormalities (25%) and diffuse ground-glass opacification which can be associated with minimal honeycombing (37%) (Table 1).2 Pathogenesis of cyst and bullae formation is unknown and it not infrequent for these findings to be confused with emphysema, especially in smokers. Despite the fact that observed lung injuries in NF1- DLD could be attributed to patient smoking behavior, as in the case of our patient, their presence in non-smokers is more plausibly due to neurofibromatosis pathogenesis itself. Pathological findings of the few cases of NF1-DLD patients who have undergone lung biopsy appear to support the hypothesis that cystic lesions are of NF1 origin rather than of smoking pathogenesis,5 owing to the presence of lymphocytic inflammation in alveolar septa, as happens in interstitial lymphoid pneumonia. Some authors argue that it could be a fibrotic environment with activation of fibroblasts and collagen production. It has also been suggested that NF1 may increase lungs sensitivity to cigarette smoking, causing early development of emphysema-like changes in these patients.6
Most common radiological findings (X-ray and chest computed tomography) in diffuse pulmonary disease associated with NF1.2,5
| Thorax X-ray | % | Chest computed tomography | % |
|---|---|---|---|
| Mottled | 32 | Ground glass | 37 |
| Linear densities | 61 | Reticular abnormality | 50 |
| Nodular densities | 13 | Bullae | 50 |
| Bullae | 73 | Emphysema | 18–25 |
| Bullae in upper localization | 65 | Cyst | 15–25 |
| Honeycombing | 12 | Honeycombing | 0 |
| Skin nodules | 39 | ||
| Subcutaneous nodules | 50 | ||
| Scoliosis | 23 | ||
| Mediastinal mass | 15 | ||
| Lung nodule | 9 |
Hitherto, no specific treatment has been approved for patients with NF1. Consequently, early diagnosis and genetic counseling are of vital importance. Current research is focused on identifying potential therapeutic targets for any of disease manifestations. Neurofibromin contains gene sequences that are similar to tumor suppressor proteins, which act as negative regulators of RAS pathway. In NF1, as neurofibromin gene mutations cause greater activity of this protooncogene (RAF/MEK and AKT/MTOR signaling pathways), with these patients experiencing increased cell replication; this would be one of the therapeutic targets currently being evaluated.7
Treatment for respiratory symptoms consists of giving up smoking and prescription of tiotropium bromide for symptomatic dyspnea relief in emphysematous patients. Corticosteroids, intended as anti-inflammatory, have not succeeded in slowing the pace of the disease progression.
In summary, when dealing with a Von Recklinghausen's patient, pulmonary involvement (NF1-DLD) should be discarded. As respiratory symptoms may be negligible, it is necessary to perform pulmonary function tests and chest CT to confirm and assess the type of injuries, as well as the extent of the disease. Smokers must give up smoking immediately; this crucial to stop disease progression. Bronchodilators may help relieve symptoms should bronchial obstruction be present.
Author's contributionsAC, NR-N and LV were responsible for the conception and design of the study, and wrote and edited the manuscript. AM-A, SC and JA contributed to the drafting and revision of the manuscript. All authors read and approved the final manuscript.
Conflicts of interestThe authors have no conflicts of interest to declare.
FundingThis study was undertaken without funding.


