Pulmonary arterial hypertension (PAH) is a progressive, fatal disease. Long-term outcomes data are scarce in Portugal. We aimed to estimate survival of newly diagnosed PAH at a Portuguese referral center in the modern management era.
MethodsBetween January 2009 and November 2015 all incident PAH cases were consecutively enrolled in a prospective cohort study. Sixty-five patients were followed up for a median of 3.1 [interquartile range 1.7–5.4] years. Kaplan–Meier survival analysis was used to estimate 1-, 3-, and 5-year survival and to compare it with a historical PAH survival estimated from the NIH cohort.
ResultsMean age was 48±19 years with female preponderance (68%). The most common PAH subgroup was congenital heart disease (PAH-CHD) (n=31; 48%), followed by connective tissue disease (PAH-CTD) (n=16; 25%), idiopathic (IPAH) (n=8; 12%) and hereditary (HPAP) (n=1; 1.5%). BNP values (hazard ratio [HR] 2.07; 95%CI 1.34–3.22; P=0.001) and male gender [HR 4.34 (1.44–13.09); P=0.009] were predictors of death. Survival rates at 1-, 3- and 5-years were 95%, 77% and 71%. Survival was not statistically different between PAH etiologies (Log-rank P=0.7). However, PAH-CHD was associated with a decreased risk of the combined endpoint of all-cause mortality and admission for decompensated heart failure [HR 0.36 (0.15–0.85); P=0.02]. We found a non-significant numerically higher survival of incident IPAH, HPAH and DPAH patients in comparison with the historical NIH cohort.
ConclusionsIn this cohort of incident PAH patients, PAH-CHD patients had better overall prognosis. Higher BNP values and male gender were associated with higher mortality.
Pulmonary arterial hypertension (PAH) is a progressive, symptomatic, and ultimately fatal disorder that involves the lung vasculature.1 Vascular proliferation and remodeling constitute the hallmark of the disease, leading to a progressive increase in pulmonary vascular resistance, right ventricular afterload and ultimately right heart failure.2 PAH is clinically divided into several groups: idiopathic (IPAH), hereditable (HPAH), drug-related (DPAH), congenital heart disease-associated (PAH-CHD), connective tissue disease-associated (PAH-CTD), portopulmonary (PoPH) and associated to infections, such as HIV or schistosomiasis.3 Pulmonary capillary hemangiomatosis (PCH) and pulmonary veno-occlusive disease (PVOD), a spectrum of diseases involving the capillaries and venules, are included in a separate group (1′) within the PAH group.3 PAH is a very rare disease with an estimated incidence of 1–3 cases per million.4 The population-based prevalence ranges from 15 to 50 per 1 million.5,6
Substantial advances in treatment have been made during the past decade.4,7,8 In the late 1980s, the National Institutes of Health (NIH) registry was the first to evaluate the epidemiology of IPAH, HPAH and DPAH during an era lacking specific PAH therapies.9 The survival rates reported at 1-, 3- and 5- years were 68%, 47% and 37%, respectively.10 In the modern management era, several registry-based studies have reported an improved survival, compared with the historical NIH registry.11
In Portugal, PAH care delivery is provided by 4 treatment centers designated by the Ministry of Health. Although retrospective12 and prospective13 short-term outcomes studies are available for Portuguese cohorts, long-term outcomes data are scarce, specifically in relation to incident patients, a critical issue to prevent immortal-time bias.11 Therefore, we aimed to characterize and estimate survival in the modern management, guideline-driven treatment era of newly diagnosed, incident PAH patients14 in a Portuguese treatment center.
MethodsBetween January 2009 and November 2015, all incident patients diagnosed with PAH were consecutively enrolled in our registry at the Pulmonary Vascular Unit of Centro Hospitalar e Universitário de Coimbra. Patients from local hospitals from the central region of Portugal, with a referral population of ca. 2.5 million, are referred to our center to undergo diagnostic workup and initiate specific therapy. We followed the Declaration of Helsinki (2008) principles, with local research ethic committee approval.
Study designWe designed a single-center, prospective cohort study based on incident cases of PAH. PAH was defined as per guidelines14: right heart catheterization (RHC)-confirmed precapillary pulmonary hypertension (PH) in the absence of other etiologies for PH (left heart disease, lung disease, chronic thromboembolic PH). Precapillary PH was defined as a mean pulmonary arterial pressure (mPAP) of ≥25mmHg and a pulmonary arterial occlusion pressure (PAOP) of ≤15mmHg. PAH was classified into six different groups based on etiology, based on the latest guidelines14: IPAH, HPAH, DPAH, PAH-CHD, PAH-CTD, PoPH, and PVOD (group 1′). Patients were ineligible if they were aged under 18 years at enrollment.
For purposes of analysis, we divided the groups into two major diagnostic subgroups: PAH-CHD patients (Eisenmenger syndrome in non-corrected systemic to pulmonary shunts and PAH after shunt correction) and non-PAH-CHD patients (IPAH, HPAH, DPAH, PAH-CTD and PoPH). IPAH, HPAP and DPAH were combined into one group to simplify survival analysis. We also stratified the population according to age and gender to perform subgroup survival analysis.
We collected information regarding demographics, clinical and laboratorial parameters at presentation, namely 6-minute walking distance (6MWD), B-type natriuretic peptide (BNP) and hemodynamics from the diagnostic, baseline RHC.
Follow-up, outcomes and exposure variablesAll follow-up visits were conducted at our institution and managed by a small group of PH specialists (G.C., R.B. and A.M.S.). Follow-up intervals and initiation of relevant therapy were determined at the physician's discretion. Because of changes in therapy, availability, and recommendations throughout the time period, treatment is registered as the latest administration on last visit.
The primary endpoint of our study was all-cause mortality. The secondary endpoint was a combination of all-cause mortality and admission for decompensated heart failure. Survival time was estimated from the date of the diagnostic RHC. At the end of the study, on November 12, 2015, vital status was obtained by access to the National Health Data Platform (Plataforma Nacional de Dados de Saúde). We also registered all hospital admissions for decompensated heart failure after the first hospital admission. No patient was lost to follow-up.
Statistical analysisContinuous variables were reported as means±SD or as medians with interquartile range (IQR) where appropriate. Categorical variables were reported as absolute frequencies and percentages. Survival analysis was performed using Kaplan–Meier curves, with the date of entry into the study defined as the date of the first diagnostic RHC or the first visit to the PH clinic for those patients that did not undergo any initial RHC. Patients that did not die were censored at the end of the study. The log-rank test was used for comparison between groups.
Univariate Cox's proportional hazards analysis was used to assess the relationship between PAH and outcomes. Co-linearity between variables was examined. Significant variables on the univariate analysis along with those variables previously reported to be related with mortality, were included in a forward stepwise multivariate Cox's proportional hazards model in order to identify independent predictors of outcomes in the overall PAH population.
We conducted three subgroup analysis, investigating the interaction between PAH etiology (PAH-CHD vs. non-PAH-CHD), age, gender and prognosis. We also conducted an exploratory analysis including the IPAH, HPAP and DPAH groups in which the actual survival was compared with the historical survival from the NIH cohort,10 using the prognostic equation as follows15: P(t)=H(t)A(x,y,z); H(t)=0.88–0.14t+0.01t2, where t=time in years; A(x,y,z)=e(0.007325x+0.0526y−0.3275z), where x=mean pulmonary artery pressure; y=mean right-sided atrial pressure; and z=cardiac index. The calculated survival was visually compared with the observed survival at the same time points with 95% CI. A P value (two-sided) below 0.05 was considered statistically significant. STATA software (STATA IC for Windows, ver. 13, Lakeway Drive, TX, USA) was used for statistical analysis.
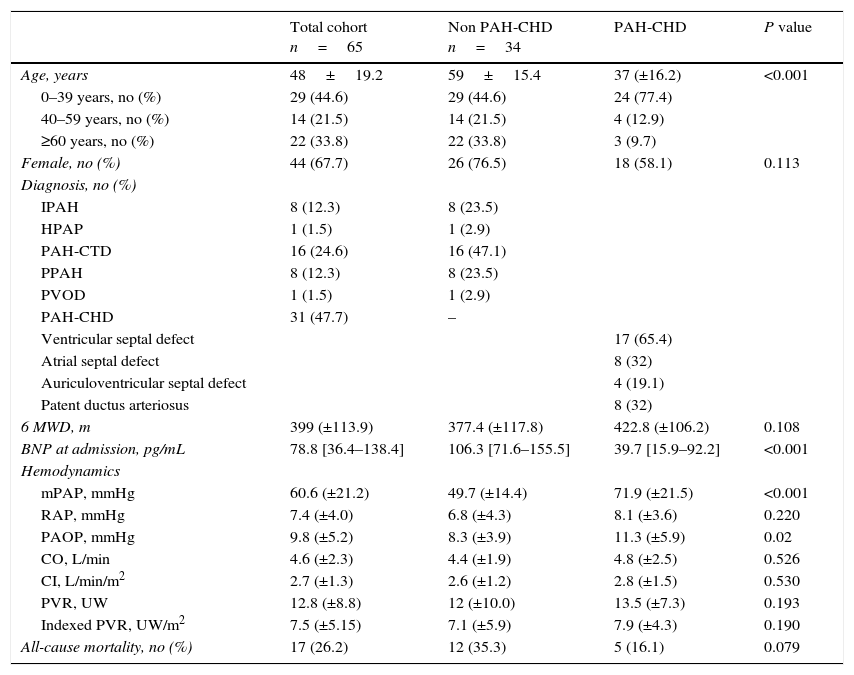
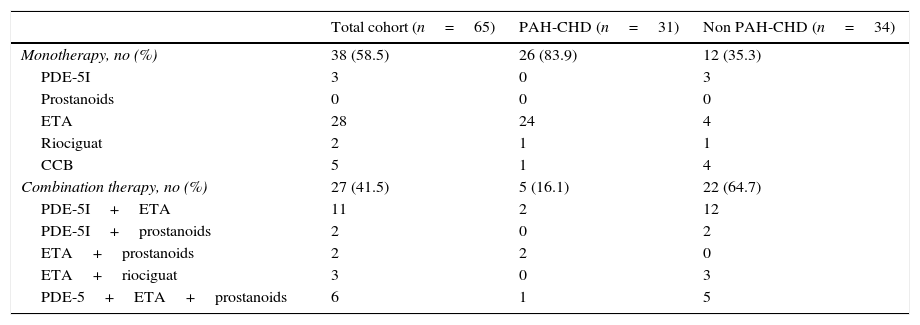
ResultsStudy populationA total of 65 patients were consecutively enrolled in our study; their clinical and hemodynamic characteristics at the time of diagnosis are shown in Table 1. The mean age was 48 (±19.2) years and the majority of patients were female (68%, n=44). The most common PAH subgroup was PAH-CHD (n=31; 48%), followed by PAH-CTD (n=16; 25%), IPAH (n=8; 12%), PoPH (n=8; 12%) and HPAH (n=1; 1.5%). On admission, the mean 6MWD was 399 (±113.9)m and the median BNP was 79 [IQR 36–138]pgdL−1. Most patients had severe hemodynamics at diagnosis, characterized by high indexed pulmonary vascular resistance (7.5±5.2Woodunitsm−2) and low cardiac index (2.7±1.3Lmin−1m−2). On the last follow-up visit, regarding pulmonary vasodilator therapy, about half (n=37) of the patients were on monotherapy and one-third (n=22) were on combination therapy (Table 2).
Clinical and hemodynamic data at the time of diagnosis. IPAH: idiopathic pulmonary arterial hypertension; HPAH: hereditary pulmonary arterial hypertension; PAH-CHD: congenital heart disease-associated pulmonary disease; PAH-CTD: connective tissue disease-associated pulmonary arterial hypertension; PPAH: porto pulmonary arterial hypertension; PVOD: veno-occlusive pulmonary disease. 6-minute walk distance; mPAP: mean pulmonary arterial pressure; RAP: right atrial pressure; PAOP: pulmonary arterial occlusion pressure; PVR: pulmonary vascular resistance.
| Total cohort n=65 | Non PAH-CHD n=34 | PAH-CHD | P value | |
|---|---|---|---|---|
| Age, years | 48±19.2 | 59±15.4 | 37 (±16.2) | <0.001 |
| 0–39 years, no (%) | 29 (44.6) | 29 (44.6) | 24 (77.4) | |
| 40–59 years, no (%) | 14 (21.5) | 14 (21.5) | 4 (12.9) | |
| ≥60 years, no (%) | 22 (33.8) | 22 (33.8) | 3 (9.7) | |
| Female, no (%) | 44 (67.7) | 26 (76.5) | 18 (58.1) | 0.113 |
| Diagnosis, no (%) | ||||
| IPAH | 8 (12.3) | 8 (23.5) | ||
| HPAP | 1 (1.5) | 1 (2.9) | ||
| PAH-CTD | 16 (24.6) | 16 (47.1) | ||
| PPAH | 8 (12.3) | 8 (23.5) | ||
| PVOD | 1 (1.5) | 1 (2.9) | ||
| PAH-CHD | 31 (47.7) | – | ||
| Ventricular septal defect | 17 (65.4) | |||
| Atrial septal defect | 8 (32) | |||
| Auriculoventricular septal defect | 4 (19.1) | |||
| Patent ductus arteriosus | 8 (32) | |||
| 6 MWD, m | 399 (±113.9) | 377.4 (±117.8) | 422.8 (±106.2) | 0.108 |
| BNP at admission, pg/mL | 78.8 [36.4–138.4] | 106.3 [71.6–155.5] | 39.7 [15.9–92.2] | <0.001 |
| Hemodynamics | ||||
| mPAP, mmHg | 60.6 (±21.2) | 49.7 (±14.4) | 71.9 (±21.5) | <0.001 |
| RAP, mmHg | 7.4 (±4.0) | 6.8 (±4.3) | 8.1 (±3.6) | 0.220 |
| PAOP, mmHg | 9.8 (±5.2) | 8.3 (±3.9) | 11.3 (±5.9) | 0.02 |
| CO, L/min | 4.6 (±2.3) | 4.4 (±1.9) | 4.8 (±2.5) | 0.526 |
| CI, L/min/m2 | 2.7 (±1.3) | 2.6 (±1.2) | 2.8 (±1.5) | 0.530 |
| PVR, UW | 12.8 (±8.8) | 12 (±10.0) | 13.5 (±7.3) | 0.193 |
| Indexed PVR, UW/m2 | 7.5 (±5.15) | 7.1 (±5.9) | 7.9 (±4.3) | 0.190 |
| All-cause mortality, no (%) | 17 (26.2) | 12 (35.3) | 5 (16.1) | 0.079 |
Medical therapy on last follow-up visit.
| Total cohort (n=65) | PAH-CHD (n=31) | Non PAH-CHD (n=34) | |
|---|---|---|---|
| Monotherapy, no (%) | 38 (58.5) | 26 (83.9) | 12 (35.3) |
| PDE-5I | 3 | 0 | 3 |
| Prostanoids | 0 | 0 | 0 |
| ETA | 28 | 24 | 4 |
| Riociguat | 2 | 1 | 1 |
| CCB | 5 | 1 | 4 |
| Combination therapy, no (%) | 27 (41.5) | 5 (16.1) | 22 (64.7) |
| PDE-5I+ETA | 11 | 2 | 12 |
| PDE-5I+prostanoids | 2 | 0 | 2 |
| ETA+prostanoids | 2 | 2 | 0 |
| ETA+riociguat | 3 | 0 | 3 |
| PDE-5+ETA+prostanoids | 6 | 1 | 5 |
CCB: calcium channel blockers; PDE-5I: phosphodiesterase 5 inhibitors; ETA: endothelin receptor antagonists.
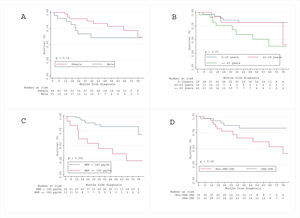
Over a median follow-up period of 3.1 [IQR 1.7–5.4] years, 17 patients (26%) died. The Kaplan–Meier survival curve for the total cohort is illustrated in Figs. 1 and 2 shows the estimated survival according to the different etiologies. Survival was numerically better in patients with PAH-CHD than in patients without PAH-CHD, although it did not achieve statistical significance (log-rank P=0.06). No significant difference was observed in estimated survival between male and female patients (log-rank P=0.07) or among different age groups (log-rank P=0.14). However, higher BNP values at diagnosis were significantly associated with increased mortality (log-rank P<0.001) (see Fig. S1 in the Supplementary Material Online).
 Global cohort of patients with pulmonary arterial hypertension (PAH) (n=65). (B) PAH cohort stratified by the different etiologies of PAH. IPAH+HPAH: idiopathic and hereditable PAH; PAH-CTD: connective tissue disease-associated PAH; PAH-CHD: congenital heart disease-associated PAH and PoPH: portopulmonary hypertension.")
Kaplan–Meier curves for all-cause mortality. (A) Global cohort of patients with pulmonary arterial hypertension (PAH) (n=65). (B) PAH cohort stratified by the different etiologies of PAH. IPAH+HPAH: idiopathic and hereditable PAH; PAH-CTD: connective tissue disease-associated PAH; PAH-CHD: congenital heart disease-associated PAH and PoPH: portopulmonary hypertension.
 (n=65).")
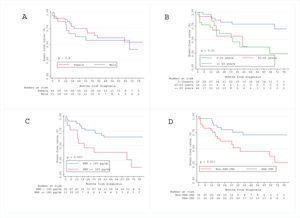
Kaplan–Meier curve for the secondary endpoint is represented in Fig. 3. Older age (log-rank P=0.010), higher BNP levels at diagnosis (log-rank P=0.003) and non-CHD-PAH (log-rank P=0.017) were associated with worse prognosis, regarding all-cause death and admission for heart failure decompensation) (see Fig. S2 in the Supplementary Material Online).
. At the 1-year time-point, there is a numerically difference of survival. At the latter time-points, the 95% confidence interval includes the observed survival. ---: observed survival; ––: predicted survival.")
Kaplan–Meier estimates with 95% confidence intervals and predicted survival using the NIH equation10,15 for the IPAH, HPAH and DPAH cohort (n=9). At the 1-year time-point, there is a numerically difference of survival. At the latter time-points, the 95% confidence interval includes the observed survival. ---: observed survival; ––: predicted survival.
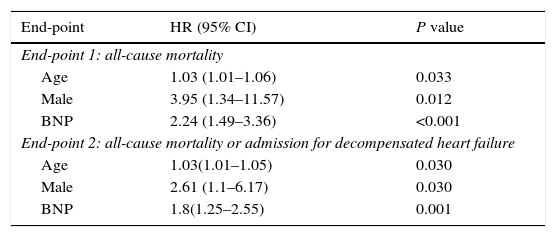
In a Cox proportional hazards model, BNP, age and male gender were independently associated with the primary and secondary endpoints (Table 3, see also Table S1 in the Supplementary Material Online). We generated and compared two subgroups from the main cohort, concerning BNP levels and gender: females with BNP levels lower than the median and males with BNP levels over the median. The rates of death [OR 8.5 (95% CI 1.4–51.5), P=0.02] and combined endpoint of death and admission for decompensated heart failure [OR 11.2 (95% CI 1.2–104), P=0.03) were significantly higher in the latter group.
Multivariate predictors of all-cause mortality and the combined endpoint of all-cause mortality and admission for decompensated heart failure in Cox's proportional hazards analysis.
| End-point | HR (95% CI) | P value |
|---|---|---|
| End-point 1: all-cause mortality | ||
| Age | 1.03 (1.01–1.06) | 0.033 |
| Male | 3.95 (1.34–11.57) | 0.012 |
| BNP | 2.24 (1.49–3.36) | <0.001 |
| End-point 2: all-cause mortality or admission for decompensated heart failure | ||
| Age | 1.03(1.01–1.05) | 0.030 |
| Male | 2.61 (1.1–6.17) | 0.030 |
| BNP | 1.8(1.25–2.55) | 0.001 |
CI denotes confidence interval, HR: hazard ratio, BNP: type B natriuretic peptide.
We conducted an exploratory analysis using the NIH prognostic equation10,15 to calculate the expected survival of our incident IPAH, DPAH and HPAH cohort at 1 and 3 years. The 1- and 3-year expected survival was 75%, and 55%, respectively; the observed survival was numerically higher (100% and 76%), although not statistically significant (Fig. 3).
DiscussionWe presented the long-term survival data of a Portuguese cohort of incident PAH patients in the modern management era. We demonstrate a 1-year survival of 95% and a 3-year survival of 77%, in line with the published series from other countries.6,16–21 The REVEAL (Registry to Evaluate Early and Long-Term PAH Disease Management) registry evaluated incident and prevalent cases of PAH and found 1- and 3-year survival rates of 85% and 68%, respectively.16 Similarly, a French multicenter registry included 121 incident PAH patients, with 1- and 3-year survival rates of 88% and 65%, respectively.17 Some smaller registries, like the Spanish and the Danish ones, yielded similar findings.6,20
Baseline disease characteristics in our cohort showed, as expected, a majority of middle-aged female patients. Similar trends were found in the French registry17 and the US REVEAL study.16 PAH-CHD was the most common form of PAH, followed by PAH-CTD. This larger proportion of PAH-CHD was not found in the REVEAL16 and French17 studies, although it was similar to the Scottish,21 Czech18 and the new Chinese registry.22
The main anomaly in our PAH population etiological distribution in regard to other published registries is the larger proportion of PAH-CHD patients (48%), a significantly higher proportion than the French17 and the REVEAL16 cohorts, somewhat higher than the Scottish (23%),21 Czech (21%)18 and similar to the Chinese registry (43%).22 PAH-CHD is associated with increased morbidity and mortality and is particularly prevalent in patients with uncorrected systemic-to-pulmonary shunts.23 Pulmonary pressures can elevate to such an extent that the shunt is reversed, resulting in the return of deoxygenated blood to the systemic circulation and the development of Eisenmenger syndrome, the most advanced form of PAH-CHD.24 Portugal lacked appropriate corrective cardiac surgery centers before the early 1980s, when most of these patients were born, therefore conditioning many patients to develop Eisenmenger syndrome. PAH-CHD patients are also clearly underrepresented in other epidemiological series, as in the French cohort due to health organization issues where many patients were followed in cardiology departments, not expert PH centers,17 and that can also explain the different prevalence of PAH-CHD among the series.
Survival in incident cohortsThe French17 and the REVEAL16 studies comprised a large percentage of prevalent cases, with a significantly worse survival for incident cases than for prevalent cases. This can be explained by inclusion of long-term survivors in the prevalent cohorts. The difference in 1-year survival between these two studies is minimal (88% vs. 85%). However, these survival rates are numerically lower than the ones in our cohort, with a 1-year survival of 95%. In terms of long-term outcomes, our cohort had a 3-year survival rate of 77%, higher than the 65–68% reported in previous studies16,17; and a 5-year survival of 71% also numerically higher than the 57% reported in the REVEAL study.16 The survival estimates presented in our population are from an incident cohort, usually displaying lower survival rates than cohorts including prevalent cases. Reasons for a higher survival rate in our cohort than in other cohorts reflect, but may be not limited to, a larger population of PAH-CHD patients, usually with better overall prognosis. Also, the availability of all drugs to treat PAH patients may help to promote survival in this group of patients.
Factors affecting survivalPAH mortality has been associated with male sex, right ventricular hemodynamic function and exercise limitation (6MWD) in the REVEAL,16 French17 and Spanish6 registries. In our study, male sex and higher BNP values were independent predictors of death and hospitalization for decompensated heart failure. Although reported in several studies, there is no clear explanation for the association of male sex and mortality.5,9,17 Additionally, no significant difference was observed in survival times between male and female patients. An improved understanding of the influence of gender on PAH outcomes is, therefore, of critical importance.5,17 Higher BNP values at diagnosis were associated with increased mortality, as also described in previous studies.25–27 This probably reflects the impact of right ventricular failure in determining the prognosis. In our registry, hemodynamic parameters did not achieve statistical significance as predictors of mortality, which can be explained by the lack of statistical power of our study population.
Study limitationsThe major limitation of our study is the small sample size, which did not allow us to identify other predictors of mortality. Our study is underpowered due to the small number of patients and events. Taking into consideration an expected mortality reduction of 43%,28 we calculated that 106 events would be necessary in order to identify a mortality reduction against placebo (i.e. the NIH patients). This number is even more limitative as we could only include IPAH, HPAH and DPAH in our NIH-comparative analysis. However, although underpowered, our analysis reflects the totality of our patients since 2009, having true value as real world data in our country. The direct comparison of the Kaplan–Meier curves of our IPAH, HPAP and DPAH subgroups against the expected survival predicted by the NIH curve has several limitations, as (i) the NIH equation was extrapolated from a mixed cohort of incident and prevalent cases, (ii) it only comprised 3 hemodynamic variables, (iii) the patients were younger with a different demographic distribution and (iv) from a cohort living 30 years ago. Although limited, this analysis is of interest as the simple observation of the curves gives a clear signal of the difference that is the actuarial survival gain with a contemporary approach. Finally, comparisons with other studies must be made with caution, given the disparity of the population size and characteristics, such as a larger cohort of PAH-CHD, which might result in overly optimistic survival estimates. The external validity of the sample is limited to centers with similar medical and pharmacological resources available.
ConclusionIn this cohort of incident PAH patients, we report a higher incidence of PAH-CHD in comparison to other PAH registries, which may need special attention. This subgroup displayed a numerically higher survival rate and overall better prognosis, compared to the non-CHD-PAH subgroup. In the global PAH population, higher BNP values and male gender were associated with higher mortality. The 1-year survival was 95% and the 3- and 5-year survival exceeded 70%, reflecting access to contemporary PAH treatment and constitute a strong incentive to the continuous work developed by all members involved in caring for this condition. Our findings encourage and reinforce the need for a long-term nationwide registry, pursuing better care for PAH patients.
Ethics approvalThis study was approved by an Ethical Committee and followed the 2008 Declaration of Helsinki principles.
Ethical responsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Authors’ contributionsP.A., conception and design, analysis and interpretation of data, manuscript writing. R.B., G.C. attending physicians, acquisition and analysis of data, revising it critically for important intellectual content. G.C., A.S., M.P. interpretation of data, revising it critically for important intellectual content. All authors read and approved the final manuscript.
Conflicts of interestThe authors have no conflicts of interest to declare.
We would like to thank Adriana Belo, MSc, for biostatistician support.