

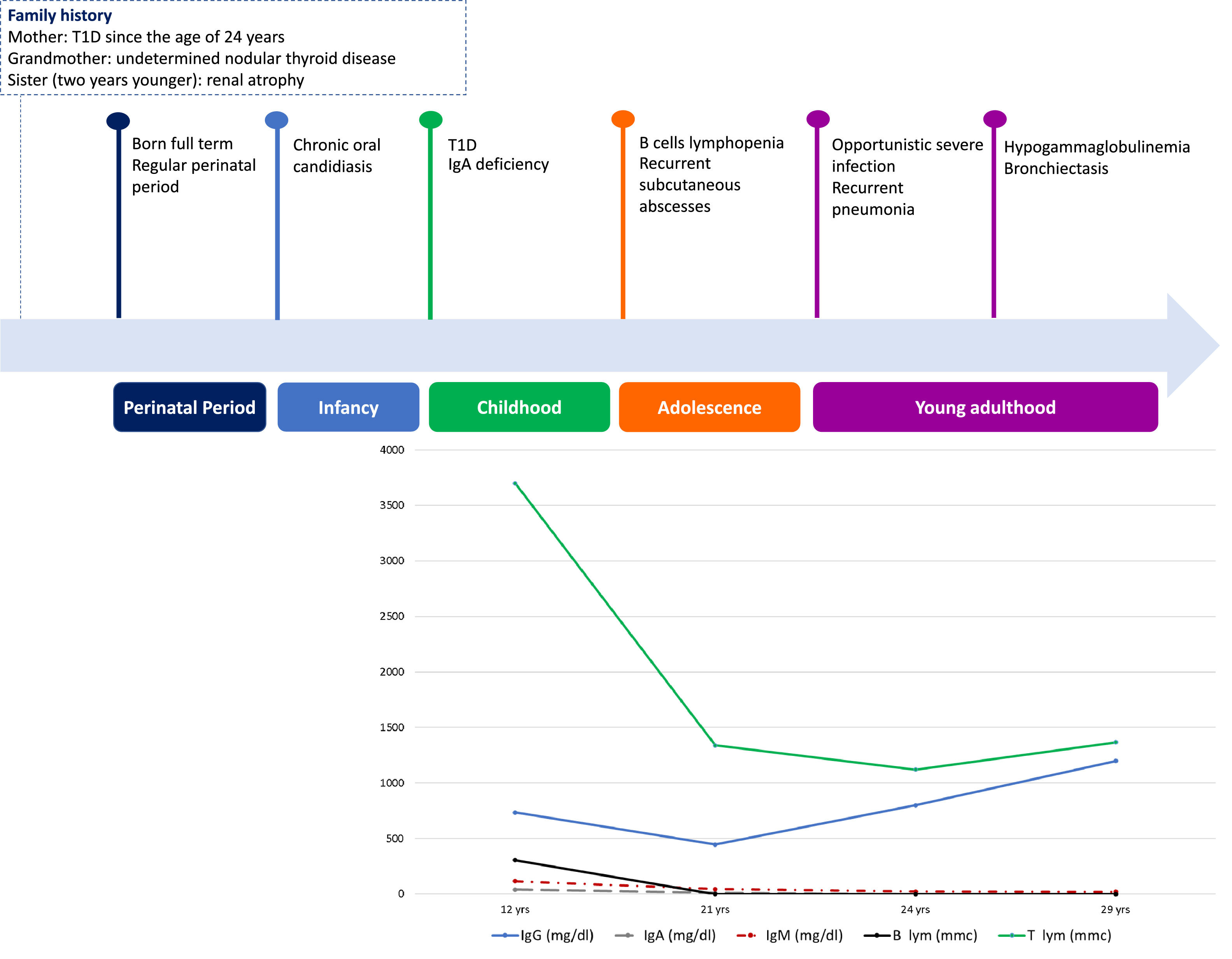
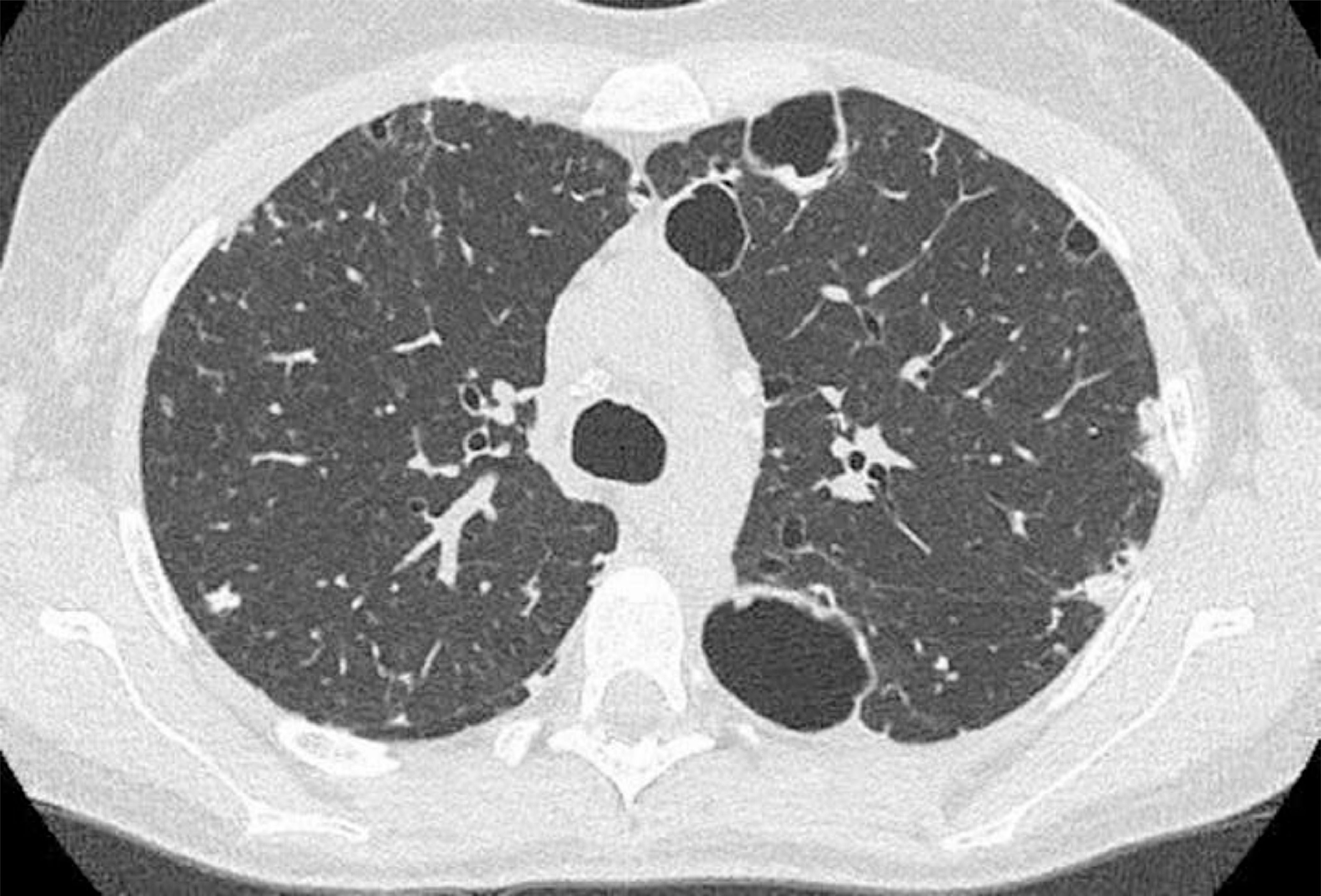
We herein describe a 29-year old female referred to our attention due to adult onset bronchiectasis and hypogammaglobulinemia. Since early infancy, she had a history of Chronic Mucocutaneous Candidiasis (CMC), followed by a diagnosis of selective immunoglobulin IgA deficiency (Fig. 1). Two years later she developed type 1 diabetes (T1D) with good metabolic control on basal-bolus insulin regimen. Type 1 autoimmune polyendocrinopathy and chronic granulomatous disease were suspected and ruled out (i.e. normal autoimmune regulator-AIRE gene expression and nitroblue tetrazolium test, respectively). She later experienced recurrent inguinal and axilla abscesses. Immunological tests were thus expanded and revealed the onset of persistent B cells lymphopenia (Fig. 1) with poor lymphocytes proliferation in response to mitogens. Isohemagglutinins and response to vaccines were normal, whereas bone marrow biopsy showed a mild dysplasia. At the age of 21-years she developed pneumonia with severe sepsis, and empirical antimicrobial therapy was started with improvement. However, chest computed tomography (CT) scan performed at 3-month follow-up showed thickened walled cavitary lesions on the right upper lobe, bilateral bronchiectasis and bronchiolitis in the upper lobes. Based on the clinical history and immune profile, common variable immunodeficiency (CVID) was diagnosed, and subcutaneous immunoglobulin (Ig) replacement therapy and co-trimoxazole prophylaxis were initiated. Despite initial favourable response, she later continued to experience intermittent cough, shortness of breath and recurrent pneumonia. At the age of 28 she was diagnosed with Pneumocystis Jiroveci and Cytomegalovirus pneumonia, documented by microscopic examination of transbronchial lung biopsy specimens and broncho-alveolar lavage fluid (BALF) PCR, respectively (Fig. 2).
 and timeline summarizing major events of the case.")
; subpleural bilateral focal consolidation centrally cavitated. Centrilobular nodules are present in the anterior segment of the right upper lobe as well. Transbronchial lung cryo biopsies documented Pneumocystis jirovecii infection.")
CT scan findings. Cysts with thickened walls in subpleural regions (the biggest are in the left lung); subpleural bilateral focal consolidation centrally cavitated. Centrilobular nodules are present in the anterior segment of the right upper lobe as well. Transbronchial lung cryo biopsies documented Pneumocystis jirovecii infection.
The patient was, thus, referred to our Unit. Immunological investigations were hence repeated and confirmed persisting B-cell lymphopenia with normal Regulatory T cells. Given the clinical history, Signal Transducer and Activator of Transcription 1 (STAT1) disorder was suspected and confirmed by targeted DNA sequencing. Namely, genetic analysis identified a rare de novo heterozygous STAT1 gene mutation (c.520T>C, p.Cys174Arg), previously reported as a pathogenic gain of function (GOF) mutation.
STAT1 plays a key role in orchestrating several pathways implicated in fundamental cellular processes, including interferons α/β signaling pathway.1 STAT1 GOF mutations lead to complex disorders characterized by great variability in the clinical manifestations. Genetic-epigenetic factors may play a role in terms of clinical expression or onset timing, and prompt diagnosis may be missed.
In the largest cohort describing 274 patients with heterozygous GOF mutations,2 the most common clinical feature was represented by fungal infections with CMC being the main manifestation at onset. Autoimmune diseases were also documented (43%), including T1D in 9%.2 The incidence of aneurysms as well as malignances affecting the skin, larynx, and gastrointestinal tract (upper and lower) was higher compared to standard population.2-4 Low Respiratory Infections were reported in almost 50%, mainly bronchitis or interstitial pneumonia. Whether or not bronchiectasis herein described is directly due to STAT1 signalling aberrance is not clear. Actually the progressive worsening of the combined immunodeficiency observed in our patient may be identified as putative factor causing the occurrence of newly acquired infections. Indeed, bronchiectasis is the ultimate outcome of many conditions due to recurrent respiratory infections.
A recent systematic review of 442 cases of STAT1 GOF mutations reported a higher risk of bronchiectasis in patients carrying the T385M mutation. However, our patients carried other type of mutation highlighting the need for proper microbiologic treatment in all patients with STAT1 GOF mutations.4
The humoral immunodeficiency herein reported was observed as a rare immunological abnormality in patients with STAT1 GOF mutations. Low B Lymphocytes or accelerated apoptosis in peripheral mature B-cells were described but the pathogenesis of impaired B cells response are still elusive.3,5
The uniqueness of the case here described resides in the challenging diagnosis due to additional clinical insights occurring over time, eventually delineating a more comprehensive phenotype culminating only in adulthood (Fig. 1). Although rare, the association of bronchiectasis in a patient displaying humoral immunodeficiency, opportunistic infections and autoimmune diseases, should prompt physicians to investigate and underlying STAT1 signalling disorder.
Indeed, early diagnosis may allow a dedicated multidisciplinary approach in order to reduce morbidity and mortality. Hematopoietic stem cell transplantation has been performed in few patients and with inconsistent results and high mortality6 and current treatment regimens are mainly supportive. However, recent novel treatment strategies targeting type I interferon signalling by systemic administration of JAK1 and JAK2 inhibitors have been successfully used in some patients.7 Regarding our patient, once the diagnosis was finalized, we referred her back to adult specialists in order to attempt the newly identified JAK1/2 inhibition therapeutical strategy.
Statement of EthicsWritten informed consent was obtained from the patient for publication of this case report.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
All authors have contributed equally to this work.


