


Severe alpha-1 antitrypsin deficiency (AATD) is generally associated with PI*ZZ genotype and less often with combinations of PI*Z, PI*S, and other rarer deficiency or null (Q0) alleles. Severe AATD predisposes patients to various diseases, including pulmonary emphysema. Presented here is a case report of a young man with COPD and AATD. The investigation of the AATD showed a novel mutation p.Leu263Pro (c.860T>C), which was named Q0gaia (Pi*ZQ0gaia). Q0gaia is associated with very low or no detectable serum concentrations of AAT.
Human alpha-1 antitrypsin (AAT), also called proteinase inhibitor (PI), belongs to the serine protease inhibitors superfamily (SERPIN) and is a mature glycoprotein; it is composed of a single chain of 394 amino acids, and three side chains of carbohydrates, weighting approximately 52 kDa. AAT is also the major SERPIN in the serum and a water soluble molecule with a half-life of 4–5 days, encoded by SERPINA1 gene located on chromosome 14.1 More than 80% of AAT is synthesized and secreted by hepatocytes, with humans producing about 34 mg/kg/day, which results in serum concentrations of 1–2 g/L. Approximately, 80% diffuses from serum into interstitial tissues and 0.5–10% reaches other biological fluids like alveolar fluid, saliva, tears, milk, semen, bile and urine.2, 3, 4SERPINA1 has numerous gene variants among common and rare alleles, and normal, deficiency and null variants, which determine protein serum levels in an autosomal codominant mode. Severe AAT deficiency (AATD) is probably one of the most common hereditary conditions, estimated to affect between 70,000 and 100,000 individuals in Europe and defined by serum AAT levels below 35% of the expected value (50 mg/dL and 11 mmol/L or 80 mg/dL, as measured by nephelometry and radial immunodiffusion, respectively).5 DAAT is generally associated with PI*ZZ genotype and less often with combinations of PI*Z, PI*S, and other rarer deficiency or null (Q0) alleles.3, 4, 6 Severe AATD predisposes patients to various diseases, including pulmonary emphysema, liver disease, systemic vasculitis and panniculitis. In Portugal the incidence of chronic obstructive pulmonary disease (COPD) is 14.2% in patients over 40 years old,7 and even though the prevalence of AATD in COPD patients is not well established, it is estimated to be about 1–3%.3 Pulmonary emphysema associated with AATD is characterized by early onset (between 35 and 45 years), which is usually accelerated by patient smoking history.
Case reportWe present a case of a 41 year old man, former smoker (18 PPY), who was referred to our pulmonology outpatient clinic in 2009, due to COPD. He had dyspnea (mMRC 2) and was treated with tiotropium bromide, formoterol, budesonide and aminophylline. The initial physical examination showed a lung auscultation with decrease breath sounds and no other significant changes. Additional exams revealed an already compromised respiratory function with very severe obstruction (FEV1/FVC 41%, FEV1 31% (1130 ml) and FVC 70%); arterial blood gas with hypoxemia (PaO2 65 mmHg) and normocapnia (35 mmHg). Furthermore, the study of alveolar capillary diffusion of carbon monoxide (DLCO) was moderately reduced (45%); chest computed tomography (CT) confirmed severe symptoms of diffuse emphysema; and AAT serum concentration was 22.2 mg/dl. Later, the evaluation of patient genotype showed incongruent results between protein electrophoresis (immunoelectrophoresis, pH 4.46 and 4.96) and protein chain reaction (PCR) multiplex screening of PI*Z and PI*S mutation,8, 9 a Z phenotype versus MZ genotype. This was explained by a novel mutation c.860T>C/p.(Leu263Pro) in exon III of SERPINA1 (genomic reference NM_000295.4; for consistency with SERPINA1 literature the amino acid was numbered according to mature protein, 24 residues less UniProtKB: P01009), predicted by different bioinformatic tools to have damaging effects in protein structure and to be a likely disease causing mutation (scores: Polyphen-2 = 1.0, SIFT = 0, PROVEAN = −5.365 and Mutation Taster = 0.99)10, 11, 12 and identified only after SERPINA1 sequencing.8 The p.Leu263Pro mutation is located in a highly conserved residue among SERPINA1 orthologs (placental mammals) and in the vicinity of p.Glu264Val (c.863A>T) variant, underlying the common deficiency S allele. Likewise, the substitution of p.Leu263Pro is likely to cause a distortion of the α-helix G and affect SERPINA1 gate structural domain. Regarding these findings, the absence of a corresponding band in protein gel electrophoresis and the patient birth place, this allele was named Q0gaia (Figure 1). Upon the diagnosis of pulmonary disease by AATD (COPD with severe airway obstruction), in a multidisciplinary meeting the patient was proposed for AAT replacement therapy. Despite the understanding of his clinical situation and the benefit he would have gained with the treatment, the patient delayed starting, due to the lack of availability for regular hospital visits. Over the past five years, the patient remained in follow-up and is currently proposed for AAT replacement therapy (60 mg/kg/week). Family screening was carried out, which identified the ZQ0 genotype in the patient's sister (a former smoker, who has FEV1 of 82%, mild reduced DLCO and no emphysema on CT chest scan).
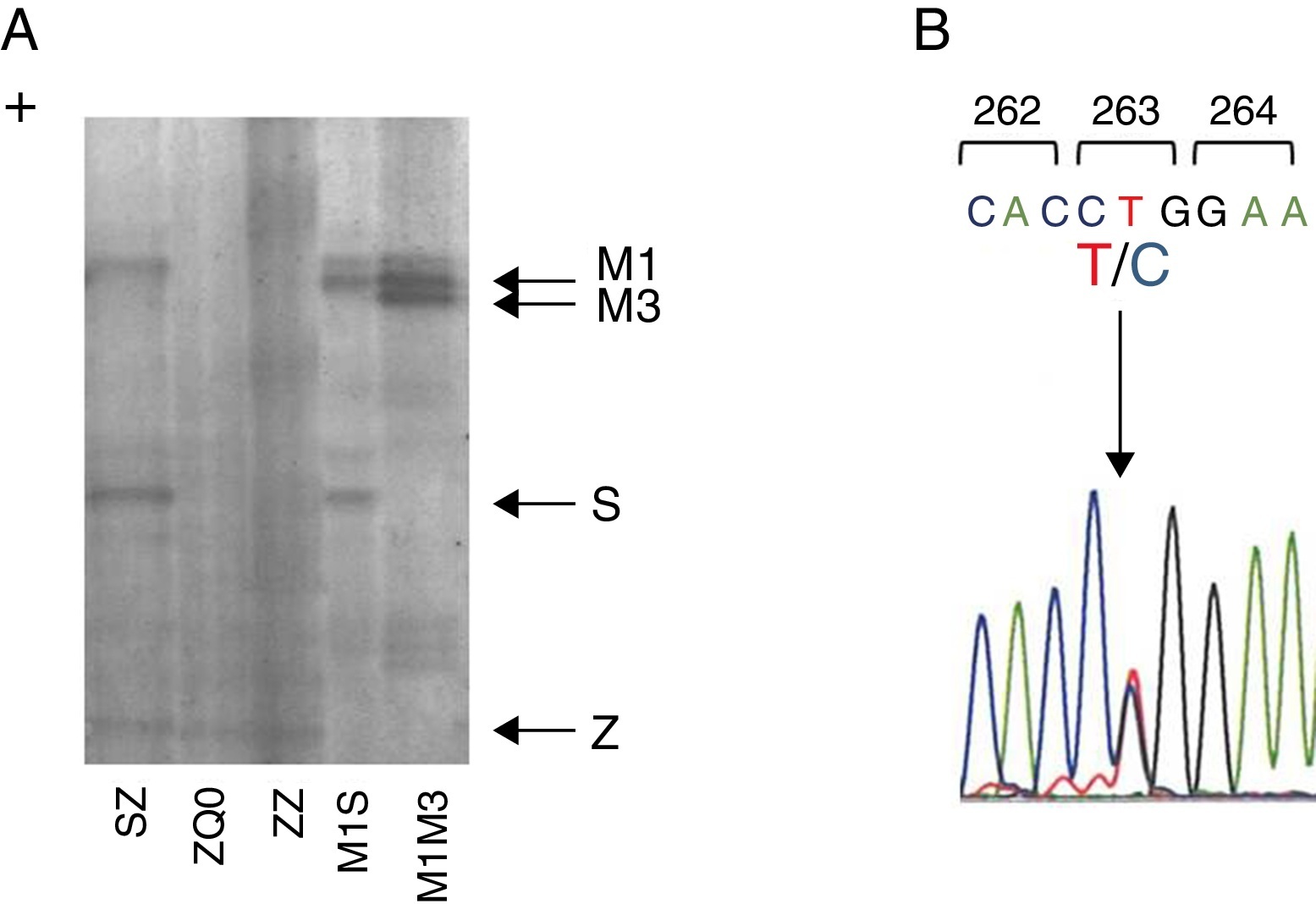
Figure 1. Characterization of Q0Gaia allele. (A) Protein gel electrophoresis. Index case ZQ0 displays only a band corresponding to PI*Z allele. (B) Electropherogram of the index case. The arrow shows the region of the T to C mutation in codon 263.
DiscussionIn this case report, COPD should probably be attributed to AATD owing to a reduced protection of the lung tissue against the neutrophil elastase, which leads to a progressive destruction of the parenchyma, and emphysema. Specifically, the increased risk of serious illness was caused by two alleles, both associated to severe AATD: PI*Z, a dysfunctional allele characterized by low secretion of AAT and an extremely rare variant Q0gaia (p.Leu263Pro) associated with very low or missing serum concentrations of AAT. Indeed, several examples from the literature show that Leu-Pro and Pro-Leu substitutions are linked to serious distortions in SERPIN structure. This is the case of SERPINA1 alleles Mprocida (p.Leu41Pro) and Mheerlen (p.Pro369Leu), which cause the distortions in A α-helix and in 1C-4B β-sheet strands, respectively, and are linked to dramatic reductions in serum levels (less than 95%), due to intracellular protein degradation.13 Here, the biochemical properties of the leucine and proline residues may correlate to mutation pathogenesis, while leucine is a hydrophobic residue displaying a preference for α-helices, the proline is a small amino acid with unique properties, able to introduce kinks in protein structure. In addition, the severe lung disease was presented in a young patient with a former history of smoking. This case report aims to demonstrate not only a novel null allele underlying AATD and its associated disease risk, but also the challenge that AAT replacement therapy may represent to the life of a patient, who takes the decision not to be treated.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.
Received 18 May 2015
Accepted 8 July 2015
Corresponding author. maria.joaooliveira@chvng.min-saude.pt


